














HTML
-
Coronaviruses(CoVs,family Coronaviridae,subfamily Coronavirinae)are important human and animal pathogens which,according to the latest release of Virus Taxonomy by the International Committee on Taxonomy of Viruses(ICTV,http://www.ictvonline.org/virusTaxonomy.asp?msl_id=26),currently comprise four distinct genera: Alphacoronavirus(αCoV),Betacoronavirus(βCoV),Gammacoronavirus(γCoV) and Deltacronavirus(δCoV). This large group of viruses has a wide spectrum of hosts,including humans,rodents,carnivores,chiropters and avians, and cause respiratory,enteric,hepatic and neurological diseases(Lai et al,2007). They include even public threats such as the severe acute respiratory syndrome(SARS) and the current Middle East respiratory syndrome(MERS)(Moratelli et al,2015). Bats are host animals of diverse αCoVs and βCoVs that may serve as the ancestral origins of mammalian CoVs(Falcon et al,2011; Woo et al,2012). In last decade,increasing numbers of bat CoVs with wide molecular diversities have been reported worldwide,particularly in China(Li et al,2005; Tang et al,2006; Woo et al,2007; Chu et al,2008; Yuan et al,2010; He et al,2014),some of which likely have the potential ability to cause human diseases(Ge et al,2013; He et al,2014; Menachery et al,2015). These findings indicate that further diverse CoVs circulate in bat populations. China has a nationwide distribution of about 120 bat species,with many roosting regions remaining uninvestigated for harbored mammal viruses. Here,we report a continuing investigation on bat-borne CoVs in some unexplored regions in China,the results of which have revealed more novel CoVs that circulate and evolve in bat populations with great molecular diversity and wide geographic distribution.
-
A total of 951 bats covering 5 families and 21 species were captured between 2005 and 2013,in Jilin,Liaoning,Yunnan,Guangdong provinces and the Tibet Autonomous Region,China. Bat species were morphologically identified by a trained field biologist and further confirmed by PCR of their mitochondrial cytochrome b gene sequence(Wang et al,2003). Respiratory and intestinal tissue specimens were collected separately from each bat and stored at –80 ℃ immediately until further processing.
-
Viral RNA of each specimen was extracted by using the RNeasy Mini Kit(Qiagen,Hilden,Germany), and was immediately reverse-transcribed with the Superscript Ⅲ Kit(Invitrogen,San Diego,CA)using r and om primers. Pan-CoV nested PCR primers were used to amplify a 440-nt sequence in the RNA-dependent RNA polymerase(RdRP)gene by our published methods(He et al. 2014)(see Supplementary Table S1 for primer information). Expected PCR amplicons were directly sequenced by the Sanger method in an ABI 3730 sequencer(Comate Bio,Changchun,China).
-
The complete RdRP genes of positive samples were amplified using LA Taq(TaKaRa,Dalian,China). Primers were designed based on RdRP gene sequences of representative αCoV and βCoV strains available in GenBank. Reactions were carried out with a touch-down PCR program: 94 ℃ for 3 min,then 10 temperature decrement cycles(94 ℃ for 30 s,58 ℃ minus 1 ℃ per cycle for 30 s,72 ℃ for 2 min),followed by 35 normal cycles(94 ℃ for 30 s,52 ℃ for 30 s,72 ℃ for 3 min), and a last extension of 72 ℃ for 10 min.
To obtain the full genomes of the interesting specimens,overlapping amplicons were obtained by the above PCR program following by assembly into contigs. In addition,deep sequencing and genome walking were also undertaken to recover more genomic sequences. The 5′ and 3′ termini were sequenced using a 5′ Full RACE Kit with TAP and a 3′ Full RACE Core Set with PrimeScript RTase(TaKaRa,Dalian,China). Primer sequences for full-length genome amplification are shown in Supplementary Table S1.
-
Genomic structures of the CoV complete sequences were predicted by the SeqBuilder program of the DNAStar software package and compared with other representatives from GenBank. Nonstructural proteins(nsps)in ORF1a and ORF1b(replicase)of the CoVs were predicted using Z-Curve version 2.0,a CoV-specific gene-finding system(Gao et al,2003).
All 400-bp amplicons(the primer truncation of 440-nt sequences)were aligned with their closest phylogenetic neighbors in GenBank using Clustal W version 2.0. The phylogenetic tree was then constructed by the maximum likelihood method of MEGA 6.06 with 1,000 bootstrap replications. To better underst and their evolutionary relationships,the complete RdRP genes were further amplified and used for the analysis.
-
The partial RdRP sequences obtained from all positive samples and the complete genome or full length RdRP sequences of some specimens were submitted to the GenBank under accession numbers KU182954 to KU183005.
Bat collection and species conformation
RNA extraction and detection by RT-PCR
RdRP gene amplification and whole genome sequencing
Genomic and phylogenetic analyses
Nucleotide sequence accession numbers
-
Of 951 bats tested 50 intestinal specimens(5.3%)were CoV positive,but surprisingly all respiratory specimens showed negative amplification. As shown in Table 1,among 181 bats from 6 species in 3 families in Guangdong province,16.2%(6/37)Rousettus leschenaultia and 27.5%(14/51)Cynopterus sphinx were CoV positive. Among 599 bats from 17 species in 5 families in Yunnan province,14.0%(14/100)Rousettus leschenaulti,2.4%(1/41)Megaerops kusnotei,9.0%(7/78)Rhinolophus sinicus and 5.3%(5/95)Myotis daubentonii were CoV positive. As the first study of this kind in the Tibet Autonomous Region,fifteen Hipposideros cineraceus and five Rhinolophus hipposideros collected in south Tibet were tested and only 6.7%(1/15)Hipposideros cineraceus showed positive amplification. In northeast China,2 of 97(2.1%)bats in Jilin province were positive: one from Murina leucogaster and another from Rhinolophus ferrumequinum. In contrast,all 16 Rhinolophus ferrumequinum and 38 Myotis ricketti in Liaoning province showed negative amplification. These results revealed a higher CoV incidence in three fruit bat species of the family Pteropodidae than in the four insectivorous bat families,indicating that fruit bats are more likely to harbor CoVs.
Bats Guangdong(Year 2005) Yunnan(Years 2012, 2013) Tibet(Year 2013) Liaoning(Year 2013) Jilin(Year 2013) Family Species Bat CoV& Bat CoV Bat CoV Bat CoV Bat CoV Pteropodidae Rousettus leschenaulti 6/37(16.2) 4 14/100(14.0) 4 Cynopterus sphinx 14/51(27.5) 4 Megaerops kusnotei 1/41(2.4) 4 Hipposideridae Hipposideros cineraceus 0/9 1/15(6.7) Hipposideros pomona 0/84 Hipposideros larvatus 0/68 0/2 Hipposideros armiger 0/11 0/18 Aselliscus stoliczkanus 0/33 Rhinolophidae Rhinolophus ferrumequinum 0/42 0/16 1/30(3.3) 2 Rhinolophus sinicus 7/78(9.0) , 2 Rhinolophus pusillus 0/5 0/6 Rhinolophus affinis 0/3 Rhinolophus hipposideros 0/37 0/5 Rhinolophus macrotis Vespertilionidae Myotis daubentonii 5/95(5.3) 3 Myotis laniger 0/8 Myotis chinensis 0/3 Myotis capaccinii 0/40 Myotis ricketti 0/38 0/27 Miniopterus schreibersi 0/8 Murina leucogaster 1/40(2.5) Megadermatidae Megaderma lyra 0/1 Note: positive/total bats; numbers in brackets indicate the coronavirus positive percentage. & CoV, α: αCoV; β: unclassified βCoV; β2: βCoV lineage 2; β3: βCoV lineage 3; β4: βCoV lineage 4. Table 1. Bat sample collection and coronavirus detection.
-
To describe the genetic relationships among the 50 sequences obtained in this study and previously known CoVs,400-nt RdRP sequences were obtained from the primer truncation of 440-nt sequences and phylogenetically analyzed. Results showed that 8 sequences grouped into 3 clusters within the genus αCoV(Figure 1A). YDB5C is the first reported bat-borne CoV(Hipposideros cineraceus)in Tibet and clustered closely with MLHJC4,a CoV from Rhinolophus sinicus in Yunnan,both sharing 94% nt identity with previously reported strain HKU2/GD/430/2006 from Guangdong(Lau et al,2007). JTAC2 identified in Murina leucogaster in Jilin province diverged considerably from known CoVs,showing the hig-hest nt identity of only 83% to bat-borne coronavirus Neixiang-14 and Neixiang-52 detected also in Murina leucogaster, and followed by 78% nt identity with some p and emic porcine epidemic diarrhea virus(PEDV)strains that have emerged recently in China,USA and Japan(Vlasova et al,2014; Sun et al,2015; Suzuki et al,2015). Five other αCoVs(MLHJC1,MLHJC6,MLHJC8,MLHJC22,MLHJC34)identified from Rhinolophus sinicus in Yunnan formed a new group with MLHJC8 being slightly more divergent,showing highest nt identities(75%–89%)with the previously reported BtCoV/860/2005(Tang et al,2006). The remaining 42 bat CoV sequences were classified as βCoV and fell into 5 clusters(Figure 1B). Twenty identified in Guangdong fell into lineage β4,which showed the geographical relationship and was further divided into two distinct clusters,one with 6 sequences sharing 99% highest nt identity with HKU9-10-1(Lau et al,2010),while another including 14 sequences sharing the closest relationship with BtCoV/BRT55630/H.lek/CK/Tha/05/2012 detected in Hipposideros lekaguli in Thail and (Wacharapluesadee et al,2015). The 21 βCoVs identified in Yunnan province exhibited considerable genetic diversity and were distributed among lineages β2,β3 and β4. Fifteen fell into β4 and further divided into 2 lineages,fourteen sequences showing closest relationship to previously reported BtCoV/BRT55629/H.lek/CK/Tha/05/2012(Wacharapluesadee et al,2015),while another(ML92C)grouped with the above Guangdong sequences. Five sequences detected from Myotis daubentonii clustered within lineage β3,sharing > 91% nt identities with previously reported HKU4-4 from Tylonycteris pachypus (Woo et al,2007). This group showed about 80% nt identity with MERS-CoVs recently identified in China(Lu et al,2015) and Korea(Kim et al,2015). The remaining Yunnan bat CoV sequence,MLHJC35,detected in Rhinolophus sinicus, and the only Jilin province sequence,JTMC15,identified in Rhinolophus ferrumequinum were clustered into β2 and showed highest nt identities to SARS-related bat-borne CoVs(SARSr-BatCoVs). MLHJC35 was 97% identical with SARSr-BatCoV Cp/Yunnan2011 previously isolated in Yunnan province(Yang et al,2011),while JTMC15 shared 99% identity with SARSr-BatCoV Rf1 found in Rhinolophus ferrumequinum, Hubei province(Li et al,2005).
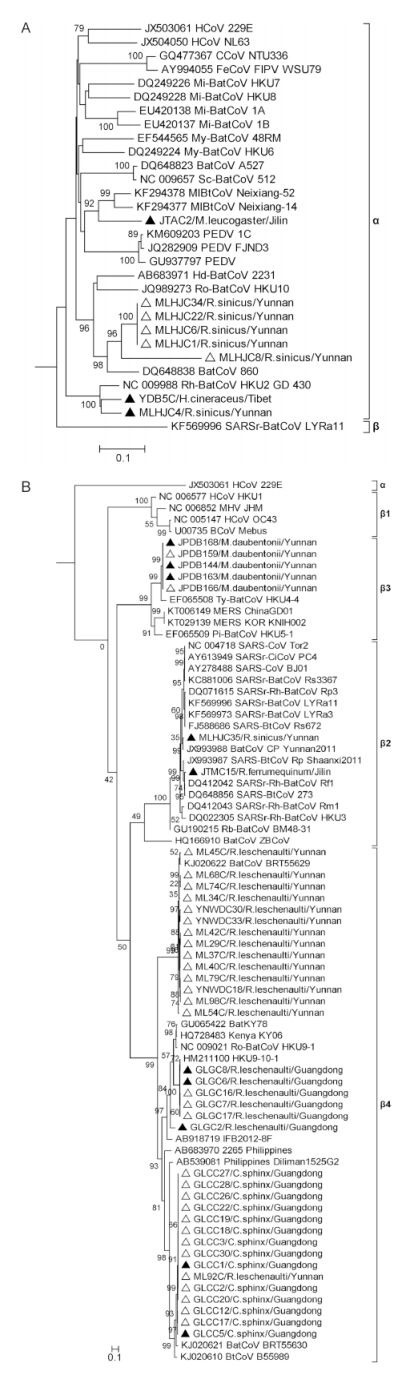
Figure 1. The 400-nt RdRP gene fragment based phylogenetic analysis of 50 bat CoV sequences obtained in this study (8 sequences of αCoV in (A) and 42 sequences of βCoV in (B)) in comparison with other representative strains retrieved from GenBank. Fifty sequences of this study are marked by triangles (13 sequences with complete RdRP gene sequencing are marked by solid triangles). The scale bar indicates the estimated number of substitutions per 10 nucleotides.
To obtain more precise analysis,representative specimens of the 8 phylogenetic clusters were subjected to full RdRP gene amplification. Complete RdRP sequences were obtained with 13 specimens belonging to 6 clusters comprised of 3 αCoVs and 10 βCoVs. Phylogenetic analysis based on the full RdRP gene sequences was highly consistent with Figure 1(Phylogenetic tree of the full RdRP gene sequences is not shown).
-
Full genomic sequencing was successful in 2 of the above 13 specimens: JTMC15 from Rhinolophus ferrumequinum, Jilin province, and JPDB144 from Myotis daubentonii, Yunnan province,with a nearly complete genome sequence obtained of JTAC2 from Murina leucogaster. The full genomes of JTMC15 and JPDB144(including complete terminal sequences of 5′ end and 3′- poly A) and near-complete genome of JTAC2 were 28,761 nt,30,321 nt and 25,719 nt in size respectively,with G+C contents of 38.1%,41.0% and 43.4%. It is proved that two proteinases,papain-like proteinase(PLPro )encoded by nsp3 gene and main proteinase(MPro )encoded by nsp5 gene in ORF1a of CoVs are able to cleave the complex of ORF1a and ORF1b(replicase)into 16 mature nonstructural proteins(nsps)(Neuman et al,2008). Our analysis of the nsps in ORF1ab revealed that all the three bat CoV genomic sequences contain 16 nsps(nsp1–nsp16)in ORF1ab,but the cleavage sites are different for nsp3 or nsp5 in different CoVs. The length of deduced amino acids of putative nsps,their first-last residue and position in replicase are shown in Supplementary Table S2.
Base on the nearly complete genomic sequence obtained,JTAC2 possesses the same genome structure as PEDVs with 7 genes in the order: 5′-ORF1a,1b,spike(S),3a,envelope(E),membrane(M) and nudeocapsid(N)-3′(Figure 2A). JTAC2 showed the nearest relationship(87.9% in ORF1a and 92.8% in ORF1b)with Lushi MI bat CoV isolates Neixiang-14 and Neixiang-52,but the latter two have very limited sequences available for further analysis. The recent PEDV-1C isolated from a piglet with diarrhea and vomiting(Sun et al,2015)was therefore used for sequence comparison and genomic organization analysis,since it has been fully sequenced and shares high identity with JTAC2(Figure 1A and Supplementary Table S3). The aa identity comparison shown in Supplementary Table S3 suggests that JTAC2 is a novel αCoV.
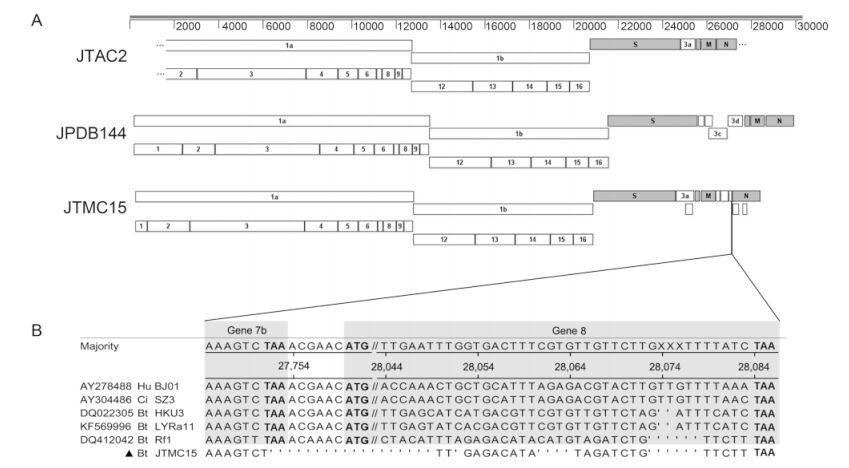
Figure 2. Predicted genome organizations of JTAC2, JTMC15 and JPDB144. (A) Nonstructural proteins are represented by open boxes, structural proteins by filled boxes. Apostrophes in JTAC2 identify unsequenced regions. (B) Sequence comparison showing the ORF shift of gene 7b of JTMC15 caused by the discontinuous deletions (represented by dots), resulting in elimination of gene 8 as compared to other SARS- and SARSr-CoVs. Nucleotide position are determined referencing strain BJ01. Stop codons and start codons are in bold fonts. Hu: human SARS-CoV; Ci: civet SARS-CoV; Bt: Bat SARS-CoV.
JTMC15 is a SARSr-BatCoV having the same genome organization as other SARSr-BatCoVs(e.g.,Rf1),but sequence deletions were observed in ORF1a and N, and between genes 7b–8. A 579-nt deletion in ORF1a of JTMC15 was also observed in SARSr-BatCoV Rs672 from a Rhinolophus sinicus bat(Yuan et al,2010) and a human SARS-CoV ShanghaiQXC2 from the late phase of the 2003 epidemic(GenBank #AY463060). This 579-nt deletion results in a 193-aa deletion of nsp3 in ORF1a,from residues 1059 to 1251 in the nucleic acid-binding(NAB)domain(Serrano et al,2009). A second deletion in the N gene of JTMC15(1156–1158 nt,one residue Q368 )was also found in 3 SARSr-BatCoV strains,Rp/Shaanxi2011(Yang et al,2013),Rm1(Li et al,2005) and 279/2005(Tang et al,2006). Interestingly,four discontinuous deletions were identified in JTMC15 between genes 7b and 8,which is unique in JTMC15,resulting in an ORF shift and elimination of gene 8(Figure 2B). Similar to known CoVs,extensive S gene variations were also observed in JTMC15,resulting in low aa identities with other SARSr-BatCoV strains(the highest being 86.1% to Rf1)as compared with other gene fragments in the genome. Receptor-binding motif(RBM)is an extended loop that lies on the surface of the receptor binding domain(RBD)of the spike protein, and is the most important domain for SARSr-BatCoV to recognize its host receptor,angiotensin-converting enzyme 2(ACE2)(Ren et al,2008; Baez-Santos et al,2015). Further alignment of the deduced amino acid sequences of RBM(55 aa)showed a closer relationship of JTMC15 to SARSr-BatCoVs than to human or civet SARS-CoVs(Supplementary Figure S1). Taking the above altogether,as shown in Figure 2A and Table 2,there are 13 genes predicted in JTMC15: 5′-ORF1a, 1b,S,3a,3b,E,M,6,7a,7b,N,9a,9b-3′. Apart from gene 7b(83.0%) and S(86.1%)all ORFs of JTMC15 had high aa identities to Rf1,ranging from 94.4%(9b gene)to 99.1%(M gene),indicating that JTMC15 is a new variant within the SARSr-BatCoV Rf1 species.
ORF JTMC15 Rf1 Rs672 BJ01 Length Length % identity Length % identity Length % identity 1a 4185 4378 98.0 4190 93.8 4383 93.5 1b 2704 2704 98.1 2704 98.1 2704 98.0 S 1236 1241 86.1 1255 81.9 1241 76.7 3a 274 274 98.2 274 92.0 274 86.2 3b 114 114 97.4 114 91.3 114 90.4 E 76 76 94.8 76 96.1 76 96.1 M 221 221 99.1 221 98.2 221 97.7 6 63 63 96.9 63 92.2 63 89.1 7a 122 122 98.4 122 93.5 122 91.9 7b 52 44 83.0 44 84.9 44 79.2 8(8a) - 122 - 121 - 39 - -(8b) - - - - - 84 - N 420 421 98.1 422 96.7 422 96.2 9a 97 97 94.9 98 79.6 98 79.6 9b 70 70 94.4 70 83.1 70 84.5 Note: # Abbreviation and accession numbers: Rf1, DQ412042; Rs672, FJ588686; BJ01, AY278488. Gene 8 in SARSr-CoVs is described as 8a and 8b in SARS-CoVs. Table 2. Comparison of ORF amino acid identities of JTMC15 and other SARS- and SARSr- CoVs#
For JPDB144,the genome organization is almost the same as HKU4-4,with 10 genes in the order: 5′-ORF1a,1b,S,3a,3b,3c,3d,E,M,N-3′(Figure 2A). However,two differences were observed in the nsp2 of JPDB144: a 12-nt insertion(residues 1143 to 1146 of 1a) and a 3-nt deletion(residue 1155 of 1a). Other JPDB144 ORFs were the same as HKU4-4 in length,sharing aa identities of between 88.8%(3c gene) and 98.8%(E gene); however,an aa sequence comparison of JPDB144 ORFs to those of HKU5 and MERS-CoV strains in the Betacoronavirus lineage 3 showed rather low similarities(Supplementary Table S4).
Detection of CoVs
Phylogenetic analysis
Full genomic sequences characterization
-
As shown in Figure 1,diverse αCoVs and βCoVs have been identified in the present study from different bats sampled at 25 locations in 4 provinces and the Tibet Autonomous Region,demonstrating the wide distribution of CoVs among a range of bat species. Of 8 αCoVs identified,YDB5C is the first bat-borne CoV identified in the Tibet and Himalayan area,detected in 1 of 15 Hipposideros cineraceus bats collected in Yadong county of Tibet,located at thesouthern edge of the Himalayas bordering on Bhutan and India. Another newly identified CoV,MLHJC4,was detected in Rhinolophus sinicus in Yunnan province,which phylogenetically clustered closely with YDB5C,both showing 94% nt identity to HKU2/GD/430/2006 identified in Guangdong(Lau et al,2007),indicating that this type of αCoV has a wide range of bat reservoirs and geo-distribution in south-west China and perhaps neighboring regions. In addition,six other αCoV sequences JTAC2,MLHJC2,MLHJC6,MLHJC8,MLHJC22 and MLHJC34,found in this study clustered as two novel CoV groups. Although not novel,the βCoVs identified here showed abundance in genetic and geographical diversities. It is interesting to note that SARSr- and MERS-like CoVs were identified,particularly JTMC15 isolated in Jilin province – the first SARSr-Bat CoV to be discovered in Northeast China.
In consideration of bat species,sampling locations and CoVs diversities,four pathogen/host/environment situations can be proposed. First,a single bat species at one location(even a single cave)harboring different CoV species(e.g.,Rhinolophus sinicus collected at the same site in Menglian county,Yunnan province,harboring three CoV species: HKU2-like,SARSr- and new αCoVs). Sec-ond,a single bat species roosting at different locations harboring the same CoVs(e.g.,Rousettus leschenaulti collected in Mengla county(south Yunnan) and W and ing county(west Yunnan)harboring the same βCoVs). Third,multiple bat species sampled at the same site harboring the same CoVs(e.g.,Rousettus leschenaulti and Megaerops kusnotei sampled at the same location in W and ing county harboring the same βCoVs). Different bat species collected at different locations may even harbor the same CoVs: e.g.,a Hipposideros cineraceus in Yadong,Tibet and a Rhinolophus sinicus in Menglian,Yunnan harbored HKU2-like viruses, and another Rhinolophus sinicus in Menglian and a Rhinolophus ferrumequinum in Tonghua county,Jilin province,harbored SARSr CoVs. Altogether,the data provided further evidence for the wide distribution of CoVs among bat populations in China, and for the suggestion that different CoVs employ different bat species as reservoirs.
The present study has identified genetically diverse bat-borne CoVs,which were detected from intestinal tissue specimens of different bat speices of wide geographic distribution. But we failed to detect any CoV sequence from the respiratory specimens that probably due to the low virus load in the lung or specific intestine-tropism of CoV in bats. Bats are considered the gene source of Alphacoronavirus and Betacoronavirus (Woo et al,2012),especially of pathogenic CoVs that cause public threats. In last decade,increasing number of CoVs have been identified in bats,in which the viral genes were presumably originated and evolved with high mutation and recombination rates(Woo et al,2007). It is apparent that large numbers of circulating CoVs remain unidentified, and are evolving worldwide within the bat population. Investigations in unexplored regions are therefore urgently needed to gain further insights into CoV diversity and evolutionary dynamics.
-
This work was supported by the Science and Technology Basic Work Program from the Ministry of Science and Technology of China (2013FY113600), NSFC-Yunnan Province Joint Fund (U1036601) and Military Medical Health (13CXZ024).
-
The authors declared that they have no conflict of interest. The whole study was approved by the Administrative Committee on Animal Welfare of the Institute of Military Veterinary, Academy of Military Medical Sciences, China (Laboratory Animal Care and Use Committee Authorization, permit number JSY-DW-2010-02). All institutional and national guidelines for the care and use of laboratory animals were followed.
-
CT conceived the study and LX carried it out with BH’s guidance. FZ, WY, TJ, GL, TH, GC, YF, YZ, QF, JF and HZ were responsible for field investigation and bat sampling. TJ and GL identified bat species morphologically. XL took part in samples screening and CoVs detection. LX wrote the paper, CT and BH then revised it. All authors read and approved the final manuscript.
Supplementary figures/tables are available on the website of Virologica Sinica: www.virosin.org;link.springer.com/journal/12250.
-
Primers Position of first nucleotide (nt) Sequences (5′3′) Pan-coronavirus nested primers CoVOF 14615 ATGGGWTGGGAYTAYCCIAARTG CoVOR 15200 TGYTGIGARCAAAAYTCRTG CoVF 14618 GGITGGGAYTAYCCIAARTGYGA CoVR 15035 CCRTCATCWGAIARWATCATCAT JTMC15 F3 2811 GCGTGTAGAYAARGTGCTTAA R3 4799 CCACGCTTRAGAAATTCAA F4 4648 GTKTCAGTDTCWTCACCAGA R4 6919 AATRCTTAACAAYAAYAGCCACAT F5 6806 CACTWCCTACRACTATWGCTAAAAAT R5 8939 GCAGARGTRGMAAARTCACTATACT F6 8827 CCTGGHTTACCDGGTACTGT R6 11329 CGYCTAGCAGCATCATCATA F10 17350 TGAGTGTYGTCAATGCTAGAC R10 19665 CTACYTTDGTGTAAACAGCATTATT F12 21283 GCTATACCATGCATGCTAACT R12 22615 CGAAAAAGARGTTGAGTTGTAG 5OR 252 ATTGGCTGAAACGACACCACTTC 5IR 161 GTCGATTAAAGCACTTGGCTCCA 3OF 27341 GACATCCCAGAGTGGAGGAG 3IF 27448 AGGTGTTGATGCCTCAGGCTAT JPDB144 F1 1 GATTTAAGWGAATAGCYTRGCTATC R1 1749 GTVGTWCCAGAVAGWARTGC F2 1572 GGTACTATGYACTTTRTKCCT R2 3846 CWGCDATRCCACCRCCAT F3 3759 GTKACHHTAGTHTTWGGTGA R3 5978 ACTAATAGYATCACYGCCA F4 5940 TAYWCTAATAGYTGCCTTG R4 8244 ACATCAGAYTCCACACC F5 796 TGGCCAGGAAARTTTAGC R5 10071 TCACTACCAGTYTCRCTGTA F6 9854 TACTGATGGTAARCTKAATTGTAG R6 12438 CATAGTTTGCATAGCACT F7 12559 TCWATGTATAAGCAAGCACGT R7 14645 GGATCWGCKGCATACATCAT F8 14556 TATCTTGTGGTTATCACTAC R8 17048 ATACCTCTCTTGATTCAC F9 16931 CGYATWGAYTATAGTGATGCTG R9 18983 ATCCCAMTCMACACGTTC F10 18791 TATGCCTGCTGGASTCATTC R10 20854 ATACTGRCACAATTGCATATATT F11 20639 CCTATTGAYTTAACWATGATTG R11 22365 GARWAGAGRTGAACRCCTTG F12 22338 GAGTGGTTYGGYATTACMCA R12 24688 GAAATAGCACCRAAAGTRTTAG F13 24267 GCWGATCCYGGYTATATGC R13 26250 CATAACGRTTKTGYYCGAAG F14 26140 ACTAAAGYATYAGCAAAACAAGA R14 27969 CGTTAAACCCASTCSTCAG F15 27842 GCTAYTMGATTATGTGTGC R15 30232 GCCTAATCTAATTGAATAATAGC 5OR 268 GTCACACTAGCCTTGGAAAGCA 5IR 83 CAGACCACAACACAACACGCACACAACA 3OF 30061 ATCATGTTARACTTACAGTGCAAG 3IF 30151 AAAGACTGTCACCTCTGCGTGATT JTAC2 F1 4010 CCACTATGTSACCAATWTYTATGAT R1 6107 CTTATCAATAAGCTTAGTAGCGTCT F2 5961 TGTYGGMCAYTATACTGTTTTTGA R2 8460 ACACGGCAATARGTCATAGC F3 8172 TGGTAAAACWCTTGTKTTTGC R3 9704 ACAAGCGCCATTAATGAA F4 9615 TTAAYATTYTGGCRTGCTATGAT R4 11457 CYTGTTCMGCCATTCTATCAA F5 11364 GTTCTCCACCTCAGTTGGT R5 13349 TCCTCACCAAAWATATCACTCTT F6 13199 GATAAYCAGGATCTTAATGGTGA R6 15458 TGACATGRTCATAAGCRCACTT F7 15309 ATTCWACTGCTAARTTTTGGGA R7 17318 CCATAAASGAKATWACATGCTCATA F8 17174 GAKGGTTGYGGTCTYTTTAAAG R8 18608 GGTGTTGTARGCATTARCATAGC F9 18470 TGCCMTTYTTYTTCTATGATG R9 20451 TCRAGCACACTRTTGTAAGACATAG F10 20301 GGACAATGTTYTGTACCAGTG R10 22672 ACATTCTTRAAGGCKARCAACTG F11 22567 AAYGTGTGCACCCAGTATACTAT R11 24958 TGAMGCTTTAAACAGTGCAA F12 24352 ATCCCAGAKTATGTYGATGTTAA R12 26127 ACCTTATAGCCYTCKACAAGCA F13 25921 CAGCATCCTTATGGCTTG R13 27429 ACTTTGGCACAGTCATYTTATAG Genome walking R1 4161 TGGCTGTAAAGTTGGCTGAGGT R2 4274 GCCACCACCATGAGACAAATTCT R3 4368 CAGAGCCAACCTTAAGTTTGCCA R4 2642 ACTTACARCTAACACCGGCCAGT R5 2745 TAGTCAAACCGTTCTCTACWGGAAT R6 2889 CGTCATAGAATGCATAACCATCAAC Table 1. Primers used in this study
nsp JTAC2$ JTMC15 JPDB144 Length (aa) First - last residuePosition Length (aa) First - last residuePosition Length (aa) First - last residuePosition 1 - - 179 M1 -G179 731 M1 -G731 2 467 Y1 -G467 639 G180 -G818 489 R732 -G1220 3 (ADRP/PLPro ) 1637 G468 -A2104 1724 A819 -G2542 1572 G1221 -A2792 4 480 G2105 -Q2584 500 K2543 -Q3042 512 T2793 -Q3304 5 (3CLPro ) 302 S2585 -Q2886 306 S3043 -Q3348 306 S3305 -Q3610 6 276 S2887 -Q3162 290 G3349 -Q3638 292 S3611 -Q3902 7 83 S3163 -Q3245 83 S3639 -Q3721 83 S3903 -Q3985 8 195 T3246 -Q3440 198 A3722 -Q3919 199 A3986 -Q4184 9 108 N3441 -Q3548 113 N3920 -Q4032 110 N4185 -Q4294 10 135 A3549 -Q3683 139 A4033 -Q4171 139 A4295 -Q4433 11 17 S3684 -D3700 13 S4172 -V4184 14 S4434 -V4447 12 (RdRP) 927 S3684 -Q4610 932 S4172- Q5103 934 S4434 -Q5367 13 (Hel) 597 S4611 -Q5207 601 A5104 -Q5704 598 A5368- Q5965 14 (ExoN) 517 A5208 -Q5724 527 A5705 -Q6231 523 S5966 -Q6488 15 (XendoU) 339 N5725 -Q6063 346 S6232 -Q6577 342 G6489 -Q6830 16 (2-O-MT) 301 S6064 -K6364 298 A6578 -N6875 302 A6831 -L7132 Note: $ The nsp2 of JTAC2 was partial sequence, lacking the 5’ terminal. Table 2. Putative nonstructural proteins (nsps) of ORF1a and ORF1b (replicase) in BatCoV JTAC2, JTMC15 and JPDB144.
ORF JTAC2 Neixiang-14 512 PEDV Length Length % identity Length % identity Length % identity 1a 3700 2030 87.9 4128 68.9 4117 76.4 1b 2680 2679 92.8 2681 85.3 2680 88.8 S 1365 - - 1371 58.6 1386 57.1 3a 224 - - 224 56.9 224 63.1 E 76 - - 76 83.1 76 83.1 M 226 - - 227 79.7 226 84.6 N 307 - - 394 62.6 441 58.8 Note: # Abbreviation and accession numbers: Neixiang-14: MIBtCoV Neixiang-14, KF294377; 512: BatCoV/512/2005, NC_009657; PEDV: PEDV-1C, KM609203. § incomplete sequences. Table 3. Comparison of ORF amino acid identities of JTAC2 with other three representative Alphacoronavirus strains#
ORF JPDB144 HKU4 HKU5 MERS Length Length % identity Length % identity Length % identity 1a 4447 4445 93.8 4481 71.1 4391 64.7 1b 2699 2699 97.7 2715 89.4 2701 87.4 S 1352 1352 94.5 1352 69.6 1353 67.1 3a (3) 91 91 89.3 121 44.8 103 46.8 3b (4a) 119 119 92.5 119 53.0 109 38.3 3c (4b) 285 285 88.8 256 39.2 246 27.8 3d (5) 227 227 93.0 223 46.6 224 46.9 E 82 82 98.8 82 80.7 82 69.9 M 219 219 97.3 220 82.3 219 84.2 N 423 423 97.9 427 74.4 413 70.8 Note: # Abbreviation and accession numbers: HKU4: BatCoV HKU4-4, EF065508; HKU5: BatCoV HKU5-1, EF065509; MERS: MERS-CoV ChinaGD01, KT006149. § ORF3a, 3b, 3c, 3d in HKU4 and HKU5 are described in MERS-CoV as ORF3, 4a, 4b and 5 respectively. Table 4. Comparison of ORF amino acid identities of JPDB144 with other two representative strains of Betacoronavirus lineage 3 (β3)# .

Figure 1. Amino acid sequences comparison of entire RBM of JTMC15, MLHJC35 (marked by asterisks) with other SARS or SARS-related coronaviruses. Middle letters: Hu: human SARS-CoV; Ci: civet SARS-CoV; Bt: Bat SARS-CoV.


 DownLoad:
DownLoad: