














HTML
-
Programmable manipulation of gene facilitates the understanding of gene function and development of genetic engineered virus vaccine (Post et al. 1990; Gasiunas et al. 2012; Jinek et al. 2012; Bull 2015; Wang H et al. 2016). Viruses, as small infectious agents, can infect all types of life forms (Koonin et al. 2006). Due to rapid evolution and diverse viral transmission strategies, viruses are still serious threats to animal health and economy (Peiris et al. 2003; Meyer and Wilke 2015; Ge et al. 2018; Zhou et al. 2018). Traditionally, editing in viral genome is based on spontaneous homologous recombination in host mammalian cells, or through lambda Red-mediated recombination at bacterial artificial chromosome (BAC) level in Escherichia coli (E. coli) (Adler et al. 2000), both of which are laborious. Therefore, efficient genome editing technologies in viral genome are still desired.
Even though the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system is employed to improve the recombination (Wang H et al. 2016), the generation of a genome-modified virus is still challenging and inconvenient due to the obstacles caused by virus. The obstacles include: (1) CRISPR/Cas9 system performed far from perfect when Cas9 protein meets rapidly increasing number of replicated viral genome DNAs; (2) manipulation in some genes could lower the replication rate of recombinant virus, making the selection task difficult; (3) reporter genes (e.g. green fluorescence protein, GFP) are still necessary for highly efficient purification of recombinant virus, which need to be removed for further study.
On the other hand, relying on lambda Red system, highly efficient homologous recombination allows simple generation of mutant virus at BAC level (Adler et al. 2000; Liu et al. 2008). The homologous sequences needed for recombination could be short as 30 bp, simplifying the preparation of donor DNA (Court et al. 2002). However, marker gene (antibiotics resistance gene) is still required for highly efficient selection. Thus, additional lambda Red-mediated recombination needs to be performed in order to remove the marker gene.
Recently, CRISPR-guided precise base-editing technologies were developed (Komor et al. 2016; Ma et al. 2016; Nishida et al. 2016; Gaudelli et al. 2017). The "base editors" (BEs) mediate conversion of C·G to T·A (Komor et al. 2016; Ma et al. 2016; Nishida et al. 2016) or A·T to G·C (Gaudelli et al. 2017) within target region without double strand breaks (DSBs). The technologies rely on deamination reaction catalysed by cytidine deaminase or adenosine deaminase rather than homology-dependent repair (HDR), therefore donor DNA is not required. Since developed, BEs were widely used in mammalian cells, animals and plants (Komor et al. 2016; Billon et al. 2017; Kim et al. 2017a; Zong et al. 2017). Recent studies reported that the third generation BE (BE3) was used to induce premature stop (iSTOP) codon with high efficiency in Staphylococcus aureus, E. coli, and Brucella melitensis (Gu et al. 2018; Zheng et al. 2018).
Pseudorabies virus (PRV, Suid alphaherpesvirus 1), which is a member of the Alphaherpesvirinae subfamily of Herpesviridae, causes devastating disease (Aujeszky's disease) in swine, resulting in great economic losses in swine agriculture (Pomeranz et al. 2005). Recently, PRV variants emerged and caused infectious disease, posing a desperate demand for efficient viral genome editing technologies to facilitate virology study and vaccine development (Wu et al. 2013). To investigate these PRV variants, several researchers have cloned PRV-BACs for use as a rapid genome editing platform (Smith and Enquist 2000; Peng et al. 2009; Wang J et al. 2016, Wang et al. 2018). We envisioned that CRISPR-guided BE would enable rapid and efficient viral genome editing at BAC level in E. coli.
In this study, we presented a highly efficient method to edit viral genes using CRISPR-guided BE3 system based on BAC. We cloned PRV-Ea genome into BAC, and manipulated viral genes in E. coli cells. The induced premature mutagenesis in US8 and UL34 showed absolutely high efficiency (100%). The US8 mutant virus was generated through reverse genetic technology and represent attenuation in intercellular transmission, which is consistent with gene function. In addition, a comprehensive bioinformatic analysis was performed to gain insight into distribution of iSTOPs in PRV variants. Taken together, our study provided a rapid and efficient approach to perform base-editing in viral genome at BAC level, which will facilitate study on virology.
-
PRV-Ea strain was isolated in 1998 and replicated in our lab (Chen et al. 1998; Liu et al. 2002; Wang et al. 2017). PK-15 and MDBK cell lines were purchased from China Center for Type Culture Collection (CCTCC) and cultured in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (Gibco).
-
The mini-F plasmid pHA2 (Adler et al. 2000) was used as F-plasmid vector, which was designed to be inserted into intergenic region between US9 and US2. For insertion, a transfer vector based on pHA2 containing homologous arms was constructed. Briefly, the upstream homologous arm (include the partial US8 gene and the entire US9 gene) and downstream homologous arm (partial US2 gene) were amplified. After digestion by Hind Ⅲ and Pac Ⅰ for amplified upstream homologous arm and digestion by Pac Ⅰ and Sac Ⅰ for amplified downstream homologous arm, the two fragments were ligated to pMD18-T digested by Hind Ⅲ and Sac Ⅰ, generating pMD18-T-US9-US2. After confirmation with digestion and Sanger sequencing, the plasmid was linearized with Pac Ⅰ and cloned into Pac Ⅰ site of pHA2, resulting in transfer vector pMD18-T-US9- US2-pHA2. The primers used are listed in Supplementary Table S1.
-
The PRV-Ea genome DNA was extracted as previously described (Wang et al. 2017). The transfer vector pMD18- T-US9-US2-pHA2 was linearized with Hind Ⅲ and purified from agarose gel. To construct an infectious PRV BAC clone, 1.5 μg of PRV-Ea genome DNA and 500 ng of purified linearized pMD18-T-US9-US2-pHA2 were cotransfected into PK-15 cells using lipofectamine 2000 (Invitrogen). Since pHA2 backbone possesses eGFP gene, the green fluorescence-positive plaques (recombinant PRV) were purified. Briefly, the plaques were picked using 100 μL tips from PK-15 cells overlaid with 1% (w/v) lowmelting point agarose and 2% FBS in DMEM. After four rounds of purification, all the plaques showed green fluorescence. A PRV-BAC recombinant was selected to infect PK-15 cells at a multiplicity of infection (MOI) of 5 to isolate the circular replicative form DNA. After 3 h, the circular replicative-form PRV-BAC DNA was isolated and eletroporated into E. coli DH10B cells (Invitrogen) with 0.1 cm cuvettes at 1.8 kV, 200Ω, and 25 μF. After recovery, the transformed cells were then plated on Luria–Bertani (LB) agar plates containing 25 μg/mL chloramphenicol and incubated at 37 ℃ till colonies grew. The colonies were cultured in 5 mL LB medium containing 25 μg/mL chloramphenicol at 37 ℃ shaking at 180 rpm for 12 h. The plasmids were extracted and confirmed by restriction fragment length polymorphism (RFLP) assay (Liu et al. 2008). Five microgram of DNA was digested with BamH I at 37 ℃ for 2 h, and electrophoresed on 0.5% (w/v) agarose gel in 1 × TAE buffer at 5 V/cm electric field. Image was captured after staining with ethidium bromide.
-
The pEcBE3 was used for base-editing in E. coli (Zheng et al. 2018). To clone sgRNA into pEcBE3, a couple of oligonucleotides were annealed and ligated into linearized pEcBE3 digested with Bsa I. The ligation products were transformed into Mach1-T1 competent cells. The plasmids were confirmed using Sanger sequencing. Oligonucleotides and primers were listed in Supplementary Table S1.
-
The DH10B carrying pPRV-BAC cells were prepared into chemically competent cells. Base-editing was performed as described previously (Zheng et al. 2018). The pEcBE3 series plasmids were transformed into DH10B carrying pPRV-BAC competent cells. After heat-shock, transformed E. coli cells were incubated in SOC medium (0.6 mmol/L IPTG) at 37 ℃ with shaking at 180 r.p.m. for 1 h, and then spread on LB agar plate (50 μg/mL ampicillin and 25 μg/mL chloromycetin). To avoid satellite colonies contamination, single colony was isolated using T-streak method. Single colony was cultured, harvest and taken for plasmid extraction. The target regions were PCR amplified and sequenced to confirm mutagenesis.
-
PK-15 cells in cell culture dish with diameter of 35 mm were transfected with 1 μg of pPRV-BAC plasmid and incubated till cytopathy occurred, thus generating rescued PRV mutants. To remove F-plasmid vector, the PK-15 cells in cell culture dish with diameter of 150 mm were transfected with 1 μg of PRV-BAC genomic DNA and 1 μg of pCAGGS-NLS/cre plasmid (Wang and Osterrieder 2011; Zou et al. 2014), and overlaid with 1.0% (w/v) low melting point agarose till PFU occurred. The rescued PRV (rPRV) was purified through plaque purification until no original PRV BAC recombinants was detected. The original PRV BAC recombinants were detected using PCR amplification using primers P11 and P12 (Supplementary Table S1).
-
Viral titers (PFU/mL) were determined by plaque assays. MDBK monolayers were infected with 100 plaque-forming units (PFU) per well in six-well plates. The cells were then overlaid with 1.6% carboxymethyl cellulose for 48 h, fixed with 10% formaldehyde, and finally stained by crystal violet. Images of discrete plaques per virus were acquired for analysis by a microscope equipped with a digital camera. Twenty-five plaques of each virus were analyzed. The average diameter for each plaque was calculated from two perpendicular diameter measurements performed using Adobe Acrobat 7.0 professional software.
-
One-step growth kinetics was conducted to compare the growth kinetics of the original strain PRV-Ea and PRVTgE mutant. PK-15 cell monolayer was infected with each virus at a MOI of 5. The cells were harvested and stored at - 80 ℃ after 0, 3, 12 and 30 h post infection (hpi). Viral titer was determined by PFU.
-
To investigate genetic stability of edited PRV infectious clone, the PRV-TgE mutant was sub-cultured for ten generation. Briefly, PK-15 cells were infected with PRV-TgE at MOI of 5. After 24 h, the cells were harvested and lysed through freezing and thawing for twice. One microliter of cell lysate was taken as template for PCR amplification and Sanger sequencing.
-
Statistical analyses were performed using Prism version 7.03 for Windows (GraphPad Software, La Jolla, CA). Values are represented as means ± standard deviations from independent experiments. Three independent experiments were performed. Statistical significance was evaluated using a two-tailed Student's t test.
-
The genome sequences of PRV-Ea, PRV-Berker, and PRVHNX strains were obtained from NCBI (GenBank accession no. KX423960.1, JF797219, and KM189912, respectively). For CAA (Gln), CAG (Gln), and CGA (Arg) codons, the distance between editable codon and PAM should be 10-14 bp; for TGG (Trp) codon, the distance should be 10-15 bp. The distribution of iSTOP codons in genome was visualized by circos (heep://circos.ca) (Krzywinski et al. 2009).
Virus Strain and Cell Line
Construction of Transfer Vector pMD18-T-US9- US2-pHA2
Construction of a PRV BAC Clone
Construction of BE3 Plasmids
Base-Editing in pPRV-BAC
Rescue and Cre-Mediated Recombination of PRV
Morphogeny Assay
Growth Kinetics Assay
Genetic Stability Assay
Statistical Analysis
Bioinformatics Analysis
-
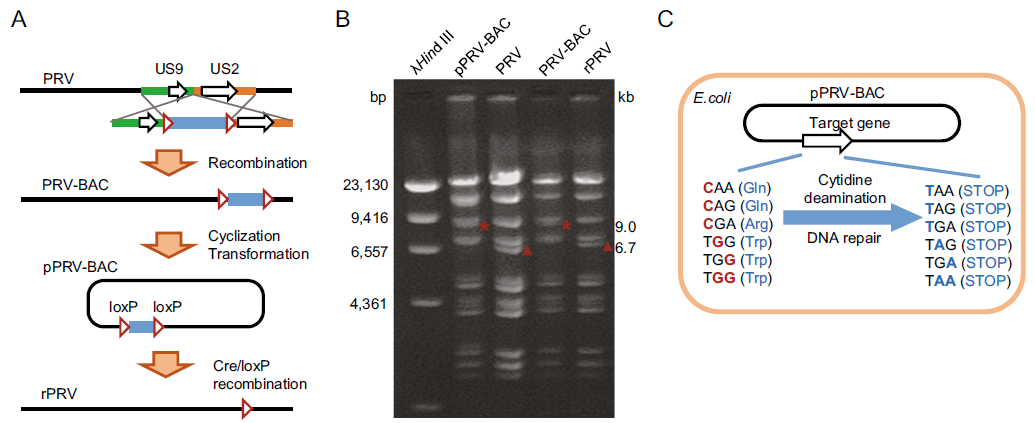
To perform base-editing in viral genome at plasmid DNA level, we designed and generated PRV-Ea infectious clone. The intergenic region between US9 and US2 gene was designed as F-plasmid vector (pHA2) integrating site. The transfer vector pMD18-T-US9-US2-pHA2 was constructed, linearized and co-transfected with PRV-Ea genome DNA into PK-15 cells, which was followed by plaque purification to obtain PRV-BAC recombinant virus. At the early infection phase, the self-cyclized PRV-BAC genome was isolated and transformed into E. coli DH10B competent cells, resulting in plasmid formed viral genome DNA (pPRV-BAC). Obviously, it is convenient to manipulate genes at plasmid level. Moreover, the F-plasmid vector was flanked by two same orientated loxP sites, thus making it simple to remove F-plasmid vector through Cre-mediated recombination (Sternberg et al. 1981) (Fig. 1A). The pPRV-BAC plasmid, wild type PRV, F-plasmid vector integrated mutant (PRV-BAC) and rescued PRV mutant (rPRV) were validated through RFLP assay with BamH I (Fig. 1B). Theoretically, the target genes in pPRV-BAC could be easily edited using CRISPR guided base-editing system in prokaryotic cells (Gu et al. 2018). The CAA, CAG, CGA and TGG codons could be converted into stop codons when cytidine deamination and DNA repair occur at the target nucleotide(s) C/G (Fig. 1C).
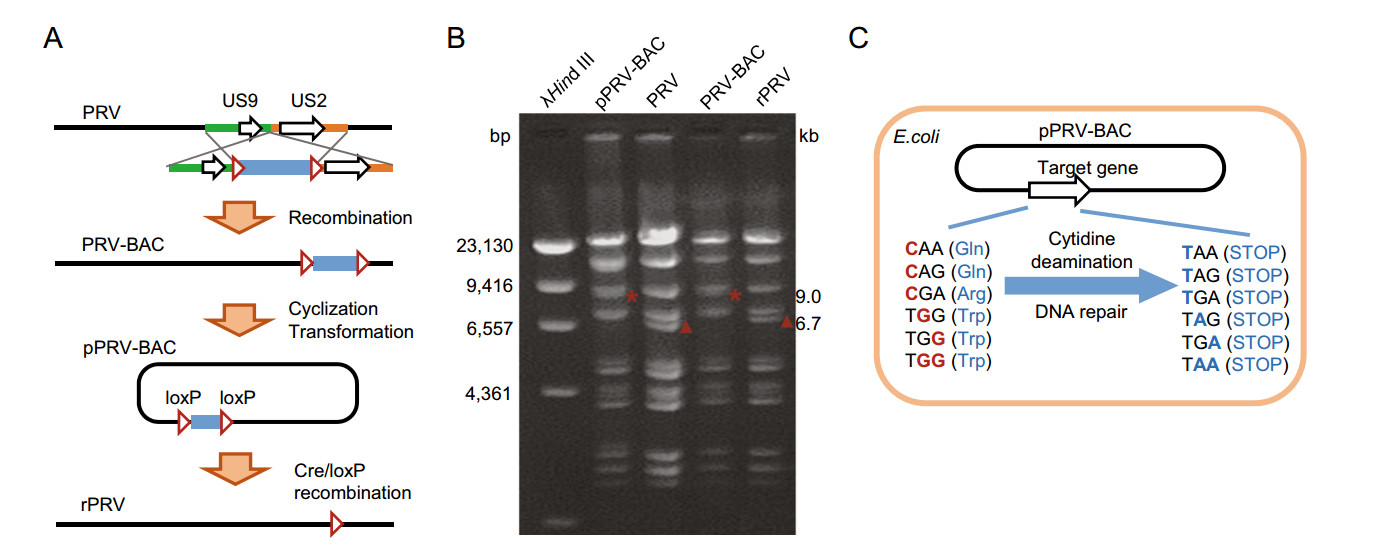
Figure 1. Schematic representation of CRISPR/Cas9 guided base-editing in viral genome based on bacterial artificial chromosome (BAC). A Schematic representation of PRV-BAC constructing process. B RFLP analysis of pPRV-BAC, PRV, PRV-BAC, and rPRV. Viral genome DNA was digested with BamH I. The affected fragments were labeled with * and ▲, which indicated 9.0 kb and 6.7 kb, respectively. C Schematic representation of induced STOP in pPRVBAC mediated by BE3 in E. coli cells.
-
To confirm the feasibility, we designed base-editing in US8 gene, which encodes 558 aa-long glycoprotein E (gE) and is involved in PRV virus cell-to-cell spread (Kratchmarov et al. 2013). After base-editing in E. coli, five colonies were cultured, harvested and taken for plasmid isolation. The US8 fragment was PCR amplified and sequenced. The Sanger sequencing results showed that all detected colonies possessed expected mutagenesis (Fig. 2A). The 46th Gln (TGG) codon was converted into iSTOP (TAA) codon, resulting in a truncated gE protein. The mutant PRV-BAC plasmid was extracted and transfected into PK-15 cells, to achieve gE truncation virus (PRV-TgE). The morphology assay showed that truncation of gE significantly suppressed PRV virus intercellular transmission, thus leading to smaller plaque size (Fig. 2B, 2C). Growth kinetic studies for PRV-TgE showed that gE is involved in virus spread between cells, but is nonessential for virus replication (Fig. 2D). The genetic stability of PRV-TgE was analyzed by passaging and Sanger sequencing. The results showed that the iSTOP mutation was stable within 10 passages (Supplementary Figure S1). In addition, we performed induced STOP at 39th and 83rd codon sites in UL34 gene that encodes 261 aa-long protein (Fig. 3A). For each site, five colonies were taken for PCR and sequencing. The editing frequencies in both designed sites were 100%. Besides the desired base conversion, synonymous mutations were carried out at the 38th, 81st, and 82nd codons (Fig. 3B). The typical editing window is position 4–8 of guide RNA target sequence (counting the end distal to PAM as position 1) in eukaryotic cells (Komor et al. 2016), whereas it could be position 2–11 in some target sites in prokaryotic cells (Zheng et al. 2018). At target site 2 in UL34, the cytosine at position 2 was also edited (Fig. 3B).
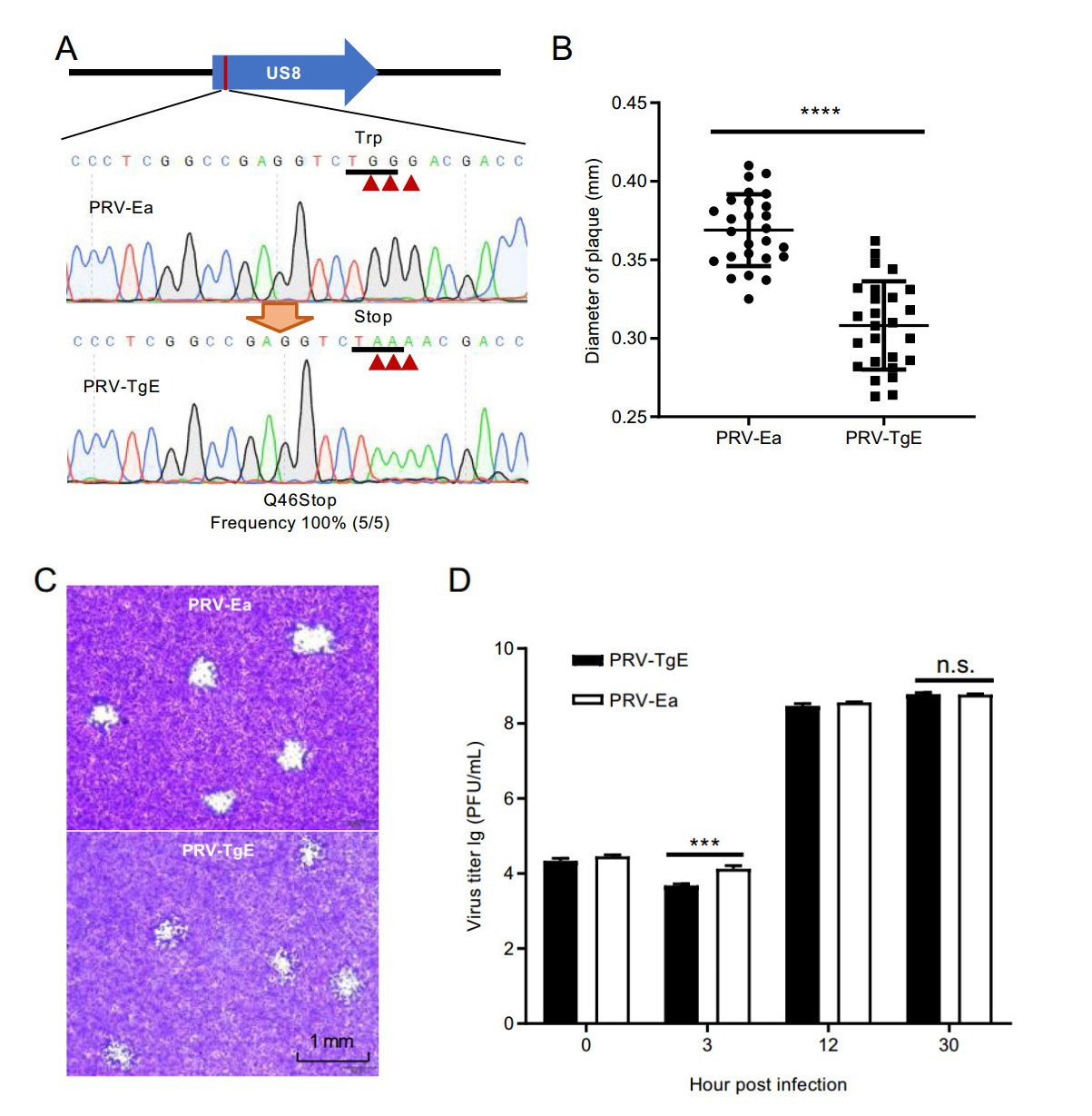
Figure 2. BE3 mediated induction of STOP in US8 gene (coding gE protein). A Sanger sequencing of BE3 induced mutagenesis in US8 gene. The substituted bases were marked with red arrows. B The impact of gE truncation on plaque size. Statistical analysis was performed with a two-way t-test. ****, P < 0.0001. C Morphogeny assay of wild type strain PRV-Ea and gE truncation strain PRV-TgE. D Growth kinetics analysis for PRV-Ea and PRV-TgE. Detailed procedure was described in Materials and Methods.
Figure 3. BE3 mediated nucleotide conversion in UL34 gene. A Scheme of designed editing sites in UL34. Protospacer adjacent motifs (PAMs) were highlighted with blue. B Sanger sequencing of BE3 induced base-editing in UL34 gene. The substituted bases were marked with red arrows.
-
Next, we performed comprehensive bioinformatic analysis to gain overall insights into targetable induced stop (iSTOP) codons of PRV-Ea genome. Besides original Cas9 formed BE3, a series BE systems were developed based on diverse CRIPSR-associated proteins, which recognized deviant PAM (Kim et al. 2017b; Li et al. 2018). These BE variants include VQE-BE3, EQR-BE3, VRER-BE3, SaBE3, SaKKH-BE3 and Cpf1-BE, which recognize NGAN, NGAG, NGCG, NNGRRT, NNNRRT and TTTN, respectively (Kim et al. 2017b; Li et al. 2018). Due to high GC content of herpesvirus genome, the Cpf1-BE preferring T-rich PAM was not taken for iSTOP codon analysis. A total of 68 ORFs were taken for iSTOP targeting scanning. Almost all ORFs (62/68) contain at least one iSTOP codon candidate (Supplementary Table S2). Among iSTOP codon candidates, 66% of candidates are located within first 20% in ORFs (Fig. 4A). About 40% (658/1643) of possibly iSTOP codons (CAA|Gln, CAG|Gln, CGA|Arg, and TGG|Trp) are targetable. The dominant potent iSTOP codons are CAGs, more than half (543/827) of which are targetable (Fig. 4B and Supplementary Table S3). Since rAPOBEC1, cytidine deaminase used in BE3 system, shows substrate bias TC C CC C AC C GC in vitro (Komor et al. 2016), we analyzed upstream base of C. The pie chart in Fig. 4B shows that 44.1% of substrates are TC and CC, indicating they are preferred and would be edited with high efficiency. In addition, CDA1-BE3 and AID-BE3, formed by cytidine deaminase homologs of rAPOBEC1, shows higher editing efficiencies over BE3 for AC and GC substrates (Komor et al. 2017). This information provides a guide for BE system selection for highly efficient editing.
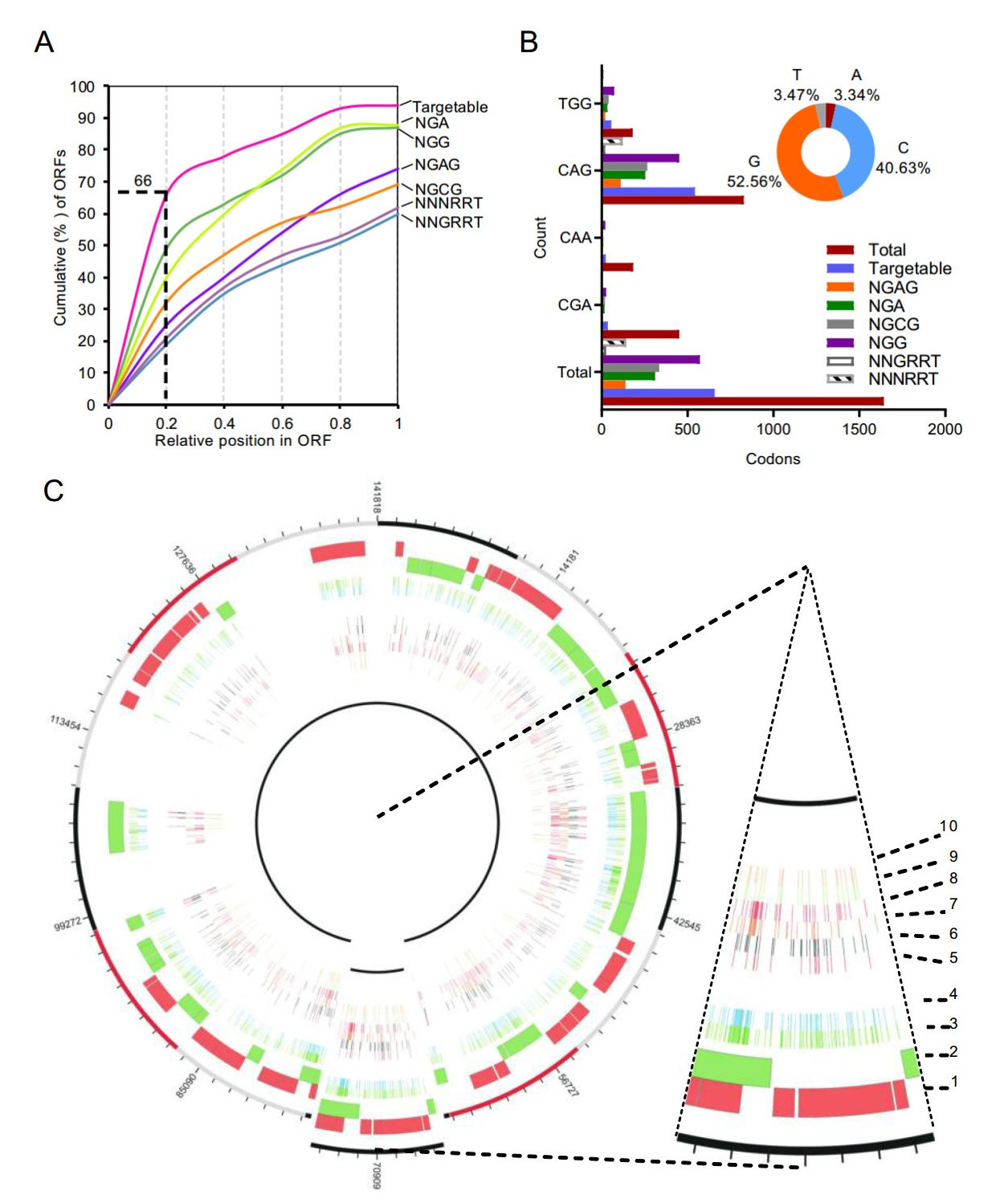
Figure 4. Comprehensive detection of iSTOP codon candidates in PRVEa genome. A Relative position of the earliest iSTOP condon candidates in PRV-Ea ORFs (cumulative percentage) by BE3 variants with distinct PAM specificities. B Overview of TGG, CAG, CAA and CGA iSTOP codon candidates targetable by BE3 variants in PRV-Ea genome. The pie chart represented upstream nucleotide distribution of substrate cytidine. C Circos plot representing the distribution of iSTOP codon candidates targetable by BE3 in PRV-Ea genome. Concentric circles from the outside to the inside: (1) open reading frames (ORFs) in forward strand of genome, (2) ORFs in reverse strand of genome; (3) total iSTOP codon candidates; (4) iSTOP codon candidates targetable by BE3 variants; (5–10) iSTOP codon candidates targetable by BE3 variants (EQE-BE3 (PAM: NGAG), VQRBE3 (PAM: NGAN), VRER-BE3 (PAM: NGCG), BE3 (PAM: NGG), SaBE3 (PAM: NNGRRT), and SaKKH-BE3 (PAM: NNNRRT), respectively).
The distribution of iSTOP codon candidates across the PRV-Ea genome is shown in Fig. 4C. We also analysed iSTOP codons in PRV-Berker strain and new-emerging PRV-HNX strain genomes. The results reveal more targetable iSTOP codons exist in both virus variants genomes (Supplementary Table S4–S5). Taken together, the bioinformatic analysis revealed that numerous coding genes in PRV genome could be truncated using BE3 system-mediated iSTOP method.
Generation of PRV-BAC
Induced Stop Mutagenesis in PRV-BAC
Prediction of Editable Sites in PRV Genome
-
In this study, we developed a highly efficient method to manipulate viral gene at bacterial artificial chromosome level. The plasmid formed infectious clone was conveniently and easily edited using CRISPR-guided base-editing system. The editing efficiency of BAC plasmid was 100%, which is consistent with editing in bacterial genome in previous study (Gu et al. 2018; Zheng et al. 2018). Because the editing is based on highly efficient base conversion mediated by CRISPR-guided cytidine deamination at plasmid, it is virus species-independent. Additionally, infectious clone is a common platform for virologists. It could be widely applied for viral genome editing by expressing BE system in E. coli strain containing infectious clone.
In previous studies, researchers used CRISPR/Cas9 system to facilitate viral gene editing. It revealed that cleavage in viral genome caused by Cas9 protein could significantly increase recombination frequency, thus improving editing efficiency (Xu et al. 2015; Yuan et al. 2015; Liang et al. 2016). Viral genome rapidly replicates in host cells, making it difficult to manipulate viral genes using CRISPR/Cas9 system in high copy number genomes. Therefore, it is essential to select appropriate method. For example, it was too difficult to isolate recombinant viruses using transfection followed by infection method. Cotransfection of CRISPR/Cas9 expression plasmid and PRV genome resulted in higher recombination rate and made it possible to obtained recombination virus (Xu et al. 2015). On the other hand, it is essential to avoid wild type virus contamination during isolation process due to viruses spread and infect cells easily. The flowcytometry based on fluorescence reporter gene simplified and facilitated isolation of recombination virus (Liang et al. 2016). Bacterial artificial chromosome method has been employed to simplify genome editing procedure, which significantly improved the recombination rate (Adler et al. 2000; Almazán et al. 2008; Liu et al. 2008; Zou et al. 2014; Feng et al. 2015; Zou et al. 2015; Richards et al. 2016; Close et al. 2017). Besides lambda Red mediated recombination (Liu et al. 2008), the CRISPR-guided base-editing method provided an alternative approach for viral gene editing on BAC platform. Our strategy could directly edit nucleotide at target loci without donor DNA and achieve absolutely high editing rate with appropriate manner. In addition, base-editing performs at BAC DNA level, indicating that the CRISPR-guided base-editing system is virus speciesindependent and plaque purification of mutant virus is not required. Our results showed that PRV-TgE mutant was stable at least 10 passages, indicating that this method might be utilized for the development of attenuated live vaccines. Previous studies demonstrated that PRV deletion mutants lacking pUL34 produced infectious progeny at 3- to 4-log-reduced titer compared to wild-type PRV (Klupp et al. 2000, 2007; Fuchs et al. 2002). Currently, we do not have UL34-expressing cell line to rescue PRV UL34 mutant. We transfected PK-15 cells with plasmid pPRV BAC harboring UL34 point mutations for three times, but failed to achieve PRV UL34 mutant. Thus, we did not analyse the plaque size and growth kinetics. Of notice, not all viral genomes have been or can be cloned as infectious plasmids. Whether the base-editing systems can be readapted for direct targeting of DNA viruses during their replication in eukaryotic cells remains to be investigated.
-
This work was supported by the National Key Research and Development Program (2016YFD0500105) and the Natural Science Foundation of China (31770191). We thank Professor Mei-Lin Jin for providing Cre expression plasmid.
-
ZFL and KZ designed the experiments; KZ performed genome editing in PRV BAC; XW and YXC constructed the PRV BAC; FFJ and LS investigated characteristic of PRV mutant; HCC provided PRV; KZ, and ZFL wrote and revised the manuscript.
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.


 DownLoad:
DownLoad: