














HTML
-
Pegiviruses are single-stranded, positive-sense RNA viruses in the family Flaviviridae. The viruses have a genome length of 9–13 kb and contain a long open reading frame that encodes a polyprotein (Smith et al. 2016; Simmonds et al. 2017). Pegivirus was previously termed GB virus and was first identified in serum samples of inoculated tamarins in 1995 (Leary et al. 1997; Simons et al. 1995). In the last few years, pegiviruses have been reported in various mammalian species, including humans, non-human primates, bats, rodents, horses, and more recently, in pigs (Baechlein et al. 2016; Berg et al. 2015; Chandriani et al. 2013; Kapoor et al. 2013a, b; Quan et al. 2013; Smith et al. 2016). Based on their genomic characterization, pegiviruses have been classified into 11 species (A–K) (Smith et al. 2016).
In 2016, porcine pegiviruses (PPgV K) were first reported in serum samples from Germany. Using nextgeneration sequencing (NGS) coupled with quantitative reverse transcription PCR (qRT-PCR) analysis, the authors revealed a 2.2% PPgV-positivity rate in domestic pigs (Baechlein et al. 2016). Subsequently, RT-PCR analysis in the United States revealed a 15.1% PPgV-positive rate, which was significantly higher than the percentages reported in other countries (Yang et al. 2018). In China, PPgV was identified in pig serum samples from four cities in the Jiangxi Province, while a complete PPgV polyprotein gene sequence was obtained from a pig farm in Guangdong Province (Lei et al. 2019). Recently, PPgV was detected in swine serum samples from different European countries, with an average PPgV-positivity rate of 2.7%, indicating that this emerging virus has a wide geographical distribution (Kennedy et al. 2019). In pigs, PPgV can cause a persistent infection that can last up to 22 months; recent studies reported a higher viral RNA load in the liver compared to that in other organs (Baechlein et al. 2016; Kennedy et al. 2019).
As PPgV is a newly discovered virus, knowledge of the epidemiology, clinical features, genetic divergence, and geographical distribution data worldwide are essential to understand the biology and genetics of this virus. In China, specifically in Guangdong Province, there are many largescale pig farms that are essential for domestic pig production. However, the prevalence and genetic characteristics of PPgV across swine herds in Guangdong Province remain unclear.
In this study, we retrospectively analyzed the presence of PPgV in swine serum samples collected between 2016 and 2018 from 20 different commercial pig farms located in nine cities across Guangdong Province. The findings reveal the wide distribution of PPgV. We also obtained two complete polyprotein gene and seven partial NS5B sequences from PPgV-positive samples and analyzed the genetic characterization of PPgV in this region.
-
From August 2016 to April 2018, a total of 339 pig serum samples were collected from 20 different large-scale pig farms located in nine cities across Guangdong Province (Zhanjiang, Shaoguan, Qingyuan, Foshan, Yangjiang, Heyuan, Yunfu, Lufeng, and Maoming). The collected samples were stored at the laboratory at -80 ℃. We collected 38, 114, and 187 samples in 2016, 2017, and 2018, respectively. No pig was sampled twice. The collected serum samples came from different pig types (sow, boar, piglet, and fattening pig) with different clinical histories. Detailed sample information, including origin, number of samples per farm, and clinical information, are provided in Table 1. Viral RNA was extracted from 200 μL of serum using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The RNA was immediately stored at -80 ℃ for further use.
Collection date District Farm ID Sample type Clinical history No. of samples No. of PPgV Positive rate Farm positive rate 2016.8 Zhanjiang A Sow serum Respiratory symptoms 14 0 0 33.3% (1/3) 2016.9 Shaoguan B Sow serum Sows abortions and stillbirths 16 1 6.25% (1/16) 2016.9 Qingyuan C Sow serum Sows abortions and stillbirths 8 0 0 2017.1 Zhanjiang D Sow serum Respiratory symptoms 30 3 10% (3/30) 33.3% (3/9) 2017.5 Foshan E Sow serum Apparently healthy 10 0 0 2017.9 Yangjiang F Sow serum Apparently healthy 13 2 15.4% (2/13) 2017.11 Shaoguan G Sow serum Apparently healthy 10 0 0 2017.11 Heyuan H Sow serum Apparently healthy 14 0 0 H Fattening pig serum Apparently healthy 2 0 0 2017.11 Qingyuan I Sow serum Sows vesicular symptoms 1 0 0 2017.11 Zhanjiang J Boar serum Apparently healthy 3 0 0 J Sow serum Apparently healthy 3 0 0 J Piglet serum Apparently healthy 9 0 0 2017.12 Foshan K Sow serum Apparently healthy 4 0 0 K Piglet serum Apparently healthy 5 1 20% (1/5) 2017.12 Heyuan L Sow serum Apparently healthy 10 0 0 2018.1 Foshan M Sow serum Apparently healthy 5 1 20% (1/5) 87.5% (7/8) 2018.1 Zhanjiang N Sow serum Sows abortions, stillbirths and diarrhea 16 0 0 2018.3 Yunfu O Sow serum Apparently healthy 10 1 10% (1/10) 2018.3 Shaoguan P Sow serum Respiratory symptoms 45 3 6.7% (3/45) 2018.3 Yunfu Q Sow serum Apparently healthy 23 1 4.3% (1/23) Q Piglet serum Apparently healthy 5 1 20% (1/5) 2018.4 Lufeng R Boar serum Apparently healthy 10 1 10% (1/10) R Sow serum Apparently healthy 25 2 8% (2/25) 2018.4 Maoming S Piglet serum Piglet diarrhea 4 1 25% (1/4) 2018.4 Heyuan T Sow serum Apparently healthy 30 1 3.3% (1/30) T Piglet serum Piglet diarrhea 14 2 14.3% (2/14) Total 20 339 21 6.2% (21/339) 55% (11/20) Table 1. Sampling results of porcine pegivirus detected with nested RT-PCR in serum samples from individual animals and herds in Guangdong Province, China in 2016–2018.
-
To screen the presence of PPgV in the serum samples, published PPgV sequences were obtained from GenBank and aligned using MegAlign software. Highly conserved sequences were found within the NS5B region and were used to design primers for nested RT-PCR (primers F0 and R0, F1 and R1; Supplementary Table S1) using Primer 6.0 software. The expected amplification products of the first primer pair (F0 and R0) and the second primer pair (F1 and R1) are 1084 bp and 870 bp, respectively. In the first round of nested RT-PCR, RNA samples were reversetranscribed to cDNA and amplified using a one-step RTPCR kit (TaKaRa Bio, Dalian, China) according to the manufacturer's instructions. The RT-PCR amplification conditions were 50 ℃ for 30 min and 94 ℃ for 2 min for the RT reaction, followed by 35 amplification cycles at 94 ℃ for 30 s, 56 ℃ for 30 s, and 72 ℃ for 1 min, with a final extension step at 72 ℃ for 7 min. In the second round, 2 μL of product from the first PCR was used as a template for PCR amplification using Premix TaqTM (TaKaRa Bio), according to the manufacturer's instructions. The PCR conditions included a pre-denaturation step at 94 ℃ for 5 min, followed by 35 amplification cycles at 94 ℃ for 30 s, 56 ℃ for 30 s, and 72 ℃ for 55 s, with a final extension step at 72 ℃ for 7 min. The second round PCR products were analyzed by 1.5% agarose gel electrophoresis.
-
A subset of PPgV-positive samples was selected for the sequencing of the polyprotein gene and partial NS5B gene. Seven primer pairs (F2–F8 and R2–R8; Supplementary Table S1) were used to amplify the complete polyprotein gene by RT-PCR using the one-step RT-PCR kit (TaKaRa Bio). The amplification program for each primer pair was 50 ℃ for 30 min and 94 ℃ for 2 min, followed by 35 amplification cycles at 94 ℃ for 30 s, 53 ℃–57 ℃ (specifi annealing temperatures for each primer pair are shown in Supplementary Table S1) for 30 s, and 72 ℃ for 80–120 s (depending on the length of the PCR product, the rate calculated was 1 kb/60 s), with a final extension step at 72 ℃ for 7 min.
The PCR products were purified using the MiniBEST DNA Extraction Kit (TaKaRa Bio) and cloned into the pMD19-T vector (TaKaRa Bio). The plasmids containing each amplicon were sequenced in both directions by Guangzhou BGI as described previously (Xie et al. 2019). Finally, two complete polyprotein gene sequences and seven partial NS5B gene sequences were obtained and deposited in the GenBank database under the following accession numbers: MH345724–MH345725 (polyprotein), and MN365881–MN365887 (NS5B) (Supplementary Table S2).
-
SeqMan software was used to splice the sequencing results. Sequence analyses of the entire polyprotein and of the NS5B and NS3 genes were performed with MegAlign and MEGA 6.06 software using the neighbor-joining (NJ) method with 1000 bootstrap replicates. The reference sequences were downloaded from the National Center of Biotechnology Information database (accessed in November 2019). The sequence of the NS3 gene was taken from our two complete polyprotein gene sequences (MH345724 and MH345725) and was used for homology and genetic evolution analyses with the sequences published in the GenBank database.
Collection of Pig Serum Samples and Viral RNA Extraction
Primer Design and Nested RT-PCR for PPgV Detection
PPgV Genome Amplification and Sequencing
Sequence Analysis
-
In this retrospective study, nested RT-PCR revealed that 21/339 (6.2%) pig serum samples from 11/20 (55%) farms were positive for PPgV (Table 1). PPgV was detected in serum samples collected as early as September 2016, indicating that the PPgV infection in Guangdong Province can be traced back to 2016 and that this emerging virus may have been circulating in the Guangdong Province for many years. The PPgV-positive rate increased from 2.6% to 7.5% between 2016 and 2018, and the positivity rates of different age groups of pigs varied from 5.2% (sows) to 13.5% (sucking piglets) (Table 2). PPgVs were detected in samples obtained from apparently healthy pigs and those with clinical symptoms, such as diarrhea, respiratory symptoms, abortions, and stillbirths (Table 1).
Sampling year Sows (%) Suckling piglets (%) Fattening pigs (%) Boars (%) Total 2016 1/38 (2.6%) – – – 1/38 (2.6%) 2017 5/95 (5.3%) 1/14 (7.1%) 0/2 (0.0%) 0/3 (0.0%) 6/114 (5.3%) 2018 9/154 (5.8%) 4/23 (17.4%) – 1/10 (10%) 14/187 (7.5%) Total 15/287 (5.2%) 5/37 (13.5%) 0/2 (0.0%) 1/13 (7.7%) 21/339 (6.2%) Table 2. The number of PPgV-positive samples of different age groups in pigs from Guangdong Province, China in 2016–2018.
-
The two complete polyprotein gene sequences obtained in this study were designated as GD1 (MH345724) and GD2 (MH345725). Sequence alignments over the complete polyprotein gene showed that the average nucleotide identity (ANI) between the two sequences was 95.2%, while the ANI with the Chinese reference sequence GDCH (MG874672) (Lei et al. 2019) was 95.1% and 97.2% for GD1 and GD2, respectively (Table 3). GD1 and GD2 had an ANI range of 95.0%–96.6% compared to four sequences identified from the USA. GD2 shared the highest ANI (96.6%) with sequence IA-22 (MG595745), whereas GD1 showed the lowest ANI (95.0%) with sequence Minnesota-1 (KY798013) (Table 3) (Yang et al. 2018; Chen et al. 2019). The alignment with the complete polyprotein gene sequences from Germany showed a low homology (87.4%–87.8% ANI). GD1 shared the highest ANI (87.8%) with sequence 903 (KU351669), whereas GD2 showed the lowest ANI (87.4%) with sequence 903 (KU351669) (Table 3) (Baechlein et al. 2016). The 8919 nucleotides of the polyprotein genes from GD1 and GD2 could be translated into 2972 amino acids. Consistent with the nucleotide homology, amino acid sequence alignment of the polyprotein gene revealed 98.8%–99.4% similarity among GD1, GD2, and strains from the USA. Whereas the sequence similarity with strains from Germany showed as low as 96.5%–96.8% (Table 3) (Baechlein et al. 2016; Chen et al. 2019; Yang et al. 2018).
Nucleotide identity (%) GenBank accession no. Strains GD1 GD2 903 80F Minnesota-1 IA-22 MO-29 ND-33 GDCH Amino acid identity (%) – 95.2 87.8 87.7 95.0 96.0 96.3 96.0 97.2 MH345724 GD1 99.0 – 87.4 87.5 95.5 96.6 95.3 95.1 95.1 MH345725 GD2 96.8 96.6 – 98.4 87.7 89.2 87.9 87.7 88.0 KU351669 903 Germany 96.7 96.5 99.6 – 87.7 89.2 87.8 87.7 87.9 KU351670 80F Germany 98.8 98.9 96.4 96.3 – 95.8 95.4 95.1 95.1 KY798013 Minnesota-1 USA 99.4 99.2 97.0 96.9 99.1 – 96.3 97.6 95.9 MG595745 IA-22 USA 99.3 99.2 96.6 96.6 99.0 99.4 – 96.4 96.2 MG595746 MO-29 USA 99.0 98.9 96.6 96.5 98.8 99.4 99.1 – 96.0 MG595747 ND-33 USA 99.3 99.0 96.5 96.4 98.8 99.2 99.1 98.9 – MG874672 GDCH China Table 3. Nucleotide identity and translated amino acid sequence identity based on the complete polyprotein gene of two sequences identified in this study (GD1 and GD2) compared with reference sequences available in GenBank.
To better understand the genomic characteristics of the PPgV sequences obtained in this study, nucleotide sequence alignment was also performed based on the partial NS3 and NS5B genes. The partial NS3 gene alignment indicated that GD1 and GD2 shared 90.7%–97.5% ANI with European sequences and 97.0%–98.8% ANI with USA sequences (Supplementary Table S3) (Chen et al. 2019; Kennedy et al. 2019; Yang et al. 2018). Nucleotide sequence alignment of the partial NS5B gene revealed that the sequences obtained in this study shared 81.9%–84.6% and 93.0%–97.1% ANI with sequences from Europe and the USA, respectively (Supplementary Table S4) (Baechlein et al. 2016; Chen et al. 2019; Yang et al. 2018). Taken together, the NS3 and NS5B alignment analyses demonstrated that PPgV sequences identified from Guangdong Province had low nucleotide homology with reference sequences from Europe, but had high nucleotide homology with sequences from the USA, which was consistent with the alignment results of the complete polyprotein gene.
-
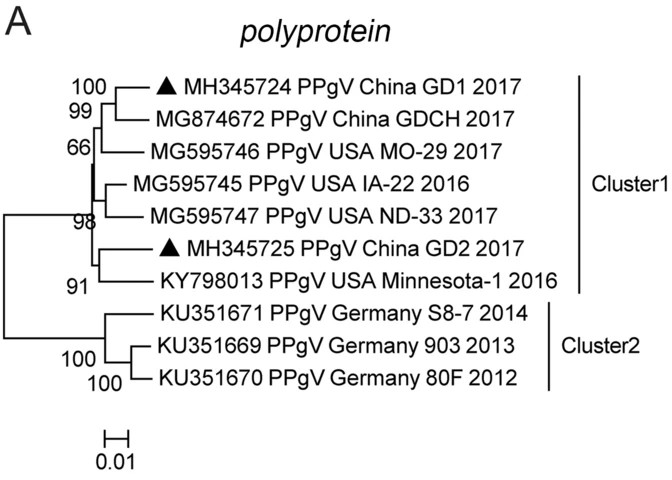
A phylogenetic tree was constructed based on the complete polyprotein gene sequences to analyze the evolutionary relationship between PPgV and other members of the Pegivirus genus. All PPgV sequences clustered independently and belonged to the pegivirus K cluster (Fig. 1). Compared to the pegivirus sequences from other species, PPgV showed a close phylogenetic relationship with bat pegivirus sequences (pegivirus B), including GB virus D strain 68 (GU566734), bat pegivirus PDB-303 (KC796073), and bat pegivirus PDB-694 (KC796083) (Fig. 1).
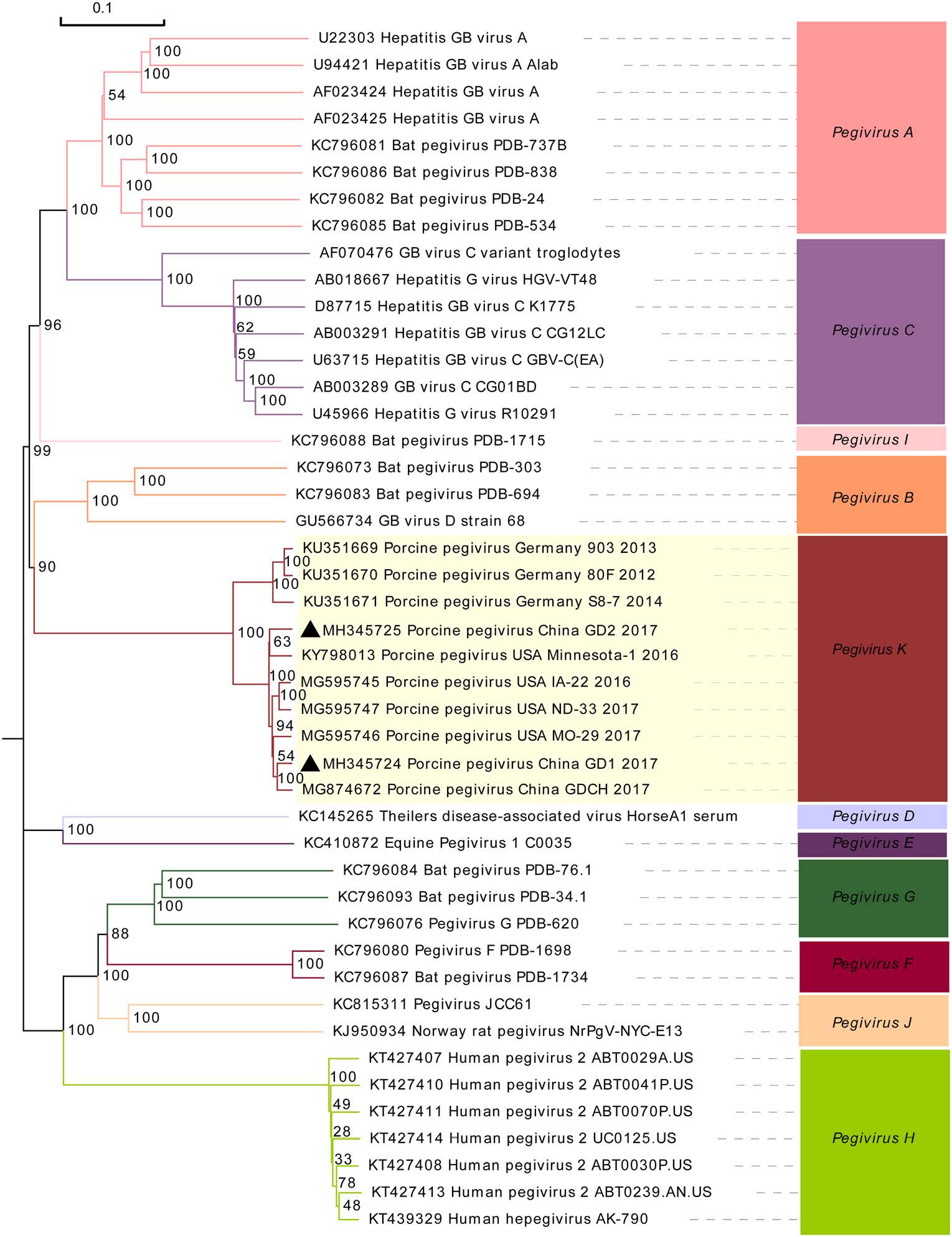
Figure 1. Phylogenetic tree based on the complete polyprotein gene of porcine pegiviruses (PPgV) and other representative pegiviruses species. Two PPgV sequences obtained in this study are marked with black triangles. MEGA 6.0 software was used to construct the neighbor-joining phylogenetic trees using a p-distance method with a bootstrap of 1000 replicates.
We plotted a phylogenetic tree based on PPgV complete polyprotein gene to investigate the evolutionary relationship between the different sequences. All the analyzed sequences belonged to two different clusters (Fig. 2A), which were subsequently analyzed and designated as cluster 1 and cluster 2. Detailed analysis of the phylogenetic tree showed that the four PPgV sequences from the USA and the only complete polyprotein gene sequence from China available before this study were in cluster 1, and sequences isolated in Germany were all in cluster 2 (Fig. 2A). The two complete polyprotein gene sequences identified in this study, GD1 and GD2, were assigned to cluster 1. GD1 formed a small sub-cluster with sequence GDCH (MG874672) and sequence ND-33 (MG595746). GD2 formed another sub-cluster in the middle of cluster 1 with the USA sequence Minnesota-1 (KY798013; Fig. 2A) (Chen et al. 2019; Lei et al. 2019; Yang et al. 2018). The phylogenetic analysis of PPgV complete polyprotein gene revealed that the two sequences identified in the Guangdong Province are closely related to sequences isolated from the USA.

Figure 2. Phylogenetic analysis based on the complete polyprotein, partial NS3, NS5B gene of PPgV. Phylogenetic tree was constructed based on analysis of PPgV strains with complete polyprotein (A), partial NS3 (1041 bp) (B), and partial NS5B (831 bp) (C) gene sequences. The PPgV sequences obtained in this study are marked with black triangles. MEGA 6.0 software was used to construct the neighbor-joining phylogenetic trees using a p-distance method with a bootstrap of 1000 replicates.
The NS3 gene contains viral helicase common motifs and is used in the phylogenetic analysis as a criterion to classify pegiviruses (Smith et al. 2016). The GenBank database contains sequences of the NS3 gene originating from six different countries. To investigate the evolutionary relationship between the sequences identified in this study and the previously published NS3 sequences, we again analyzed the phylogenetic tree (Fig. 2B). All the analyzed NS3 sequences were divided into two large clusters (cluster 1 and cluster 2). Cluster 1 was further divided into the 1.1 and 1.2 sub-clusters, and cluster 2 into the 2.1 and 2.2 sub-clusters (Fig. 2B). The phylogenetic tree showed that sequences from different countries intertwined to form different clusters, particularly the sequences identified from Europe, which were present in almost every sub-cluster, suggesting that PPgV evolution in Europe is very complex (Fig. 2B). The two sequences identified in this study were located in sub-cluster 1.1. GD2 formed a small branch with two sequences from the USA and GD1 formed another small branch with sequence GDCH (MG874672), similar to the phylogenetic tree constructed using the complete polyprotein gene.
Similar to the NS3 gene, the NS5B gene is commonly used in the phylogenetic analysis as a criterion to classify pegiviruses (Smith et al. 2016). To explore the evolutionary relationship between NS5B sequences identified in this study and previously published PPgV NS5B sequences, a phylogenetic tree analysis based on the partial NS5B gene was also performed (Fig. 2C). All sequences were in cluster 1 and cluster 2, with all nine of the presently isolated sequences present in cluster 1. More specifically, five sequences from this study and the USA sequence Minnesota-1 (KY798013) were located in sub-cluster 1.2, and the sequences of the remaining four were localized to subcluster 1.1 together with three other USA sequences (Fig. 2C). This result was consistent with the phylogenetic analysis of the complete polyprotein gene and the NS3 gene, indicating that sequences in cluster 1 are prevalent in Guangdong Province, with a high evolutionary similarity to sequences identified in the USA.
-
Analysis of NS3 amino acid mutations is essential to understand the molecular features of different PPgV strains. Based on the amino acid alignment of the available NS3 sequences (347 amino acids), we identified 11 mutation sites with no deletion or insertion mutations (Fig. 3). Moreover, there was no subgroup-specific mutation in the NS3 region. The three most frequent mutated positions were glycine in position 139, which was mutated to serine in nine strains; isoleucine in position 329, which was mutated to valine in eight strains; and alanine in position 197, which was mutated to threonine, proline, or serine in six strains (Fig. 3). A total of 14 strains harbored amino acid mutations in the NS3 gene (Fig. 3). Strain MH979672 from Poland contained five mutated sites (P127S, G139S, A197T, I329V, and F337Y). Two strains (MH979660 and KU351671) from the UK and Germany contained three mutated sites. The Chinese strain MH979668 and the American strain MG595740 contained one amino acid mutation site (I329V and A197T, respectively). The remaining nine strains contained two amino acid mutation sites (Fig. 3). Alignment of the amino acid sequences demonstrated that strains from Poland, Germany, and the UK contained more amino acid mutation sites, while those from China and the USA contained fewer mutations. Further studies are needed to determine the effect of these mutations on the viral protein structure and function.
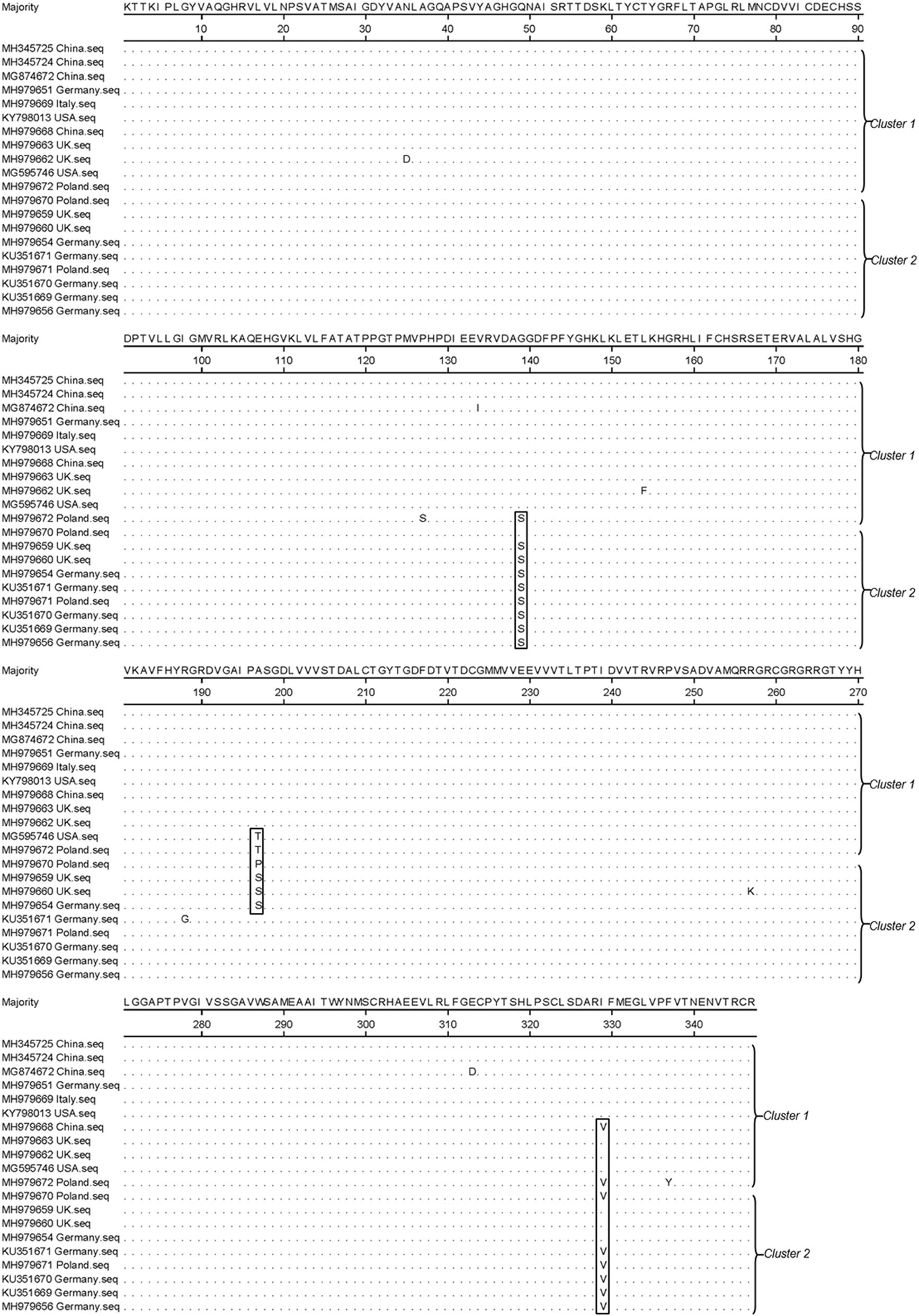
Figure 3. Alignment of deduced amino acid sequences of PPgV NS3 gene (partial, 347 amino acids). Three amino acid mutation sites (G139S, A197T/P/S, and I329V) are depicted using rectangular boxes.
Epidemiological Characteristics of PPgV in Guangdong Province
Alignment Analysis of the Full Polyprotein, and Partial NS3 and NS5B Gene Sequences
Phylogenetic Analysis of PPgV Strains GD1 and GD2
Mutation Analysis of Amino Acid Sequences of the NS3 Region
-
PPgV, also known as Pegivirus K, belongs to the genus Pegivirus, family Flaviviridae (Kennedy et al. 2019; Smith et al. 2016). PPgV was first discovered in swine serum samples in Germany (Baechlein et al. 2016). Subsequent studies confirmed the detection of this emerging swine virus in pig herds from Germany, USA, China, Poland, Italy, and the UK (Chen et al. 2019; Kennedy et al. 2019; Lei et al. 2019; Yang et al. 2018). In this study, we investigated the presence of PPgV in 339 serum samples collected between 2016 and 2018 from 20 different pig farms in nine cities across Guangdong Province. The average PPgV detection rate in pigs and swine farms was 6.2% and 55%, respectively. The percentage of PPgV-positive pigs in Guangdong Province was slightly higher than that observed in Germany (2.2%) but lower than that reported in the USA (15.1%) (Baechlein et al. 2016; Yang et al. 2018). Notably, the PPgV-positive rate increased gradually over the three years in this study. The PPgV-positive rate of 87.5% (7/8) in 2018 indicated a relatively high level of PPgV infection across Guangdong Province (Table 2). Therefore, PPgV infection in Guangdong must be taken seriously.
Human pegiviruses (HPgV) are one of the most prevalent human RNA viruses and have been reported to cause coinfection with hepatitis C virus and human immunodeficiency virus (Baechlein et al. 2016; Chen et al. 2019; Kennedy et al. 2019). Some studies have shown that HPgV could be associated with unexplained encephalitis, hemophilia, lymphoma, and febrile illness (Chen et al. 2019; Kennedy et al. 2019; Yang et al. 2018). Pegivirus D has been identified as the etiological agent of Theiler's disease in horses and is also known as acute equine hepatitis disease (Chandriani et al. 2013; Chen et al. 2019; Yang et al. 2018). Currently, the pathogenicity of PPgV or its association with other diseases remains unclear as Koch's postulates have not been established for this virus. Recent studies have shown that PPgV infection is very complicated from a clinical point of view. Yang et al. (2018) performed next-generation sequencing using serum samples from swine with vesicular disease to detect potential viral infections. The samples were negative for the Seneca Valley virus and foot-and-mouth disease virus, but positive for PPgV (Yang et al. 2018). Chen et al. (2019) performed next-generation sequencing on pig serum samples obtained from farms experiencing an increase in the incidence of mummified and stillborn fetuses. PPgV was the only virus detected. Kennedy et al. (2019) reported that porcine liver tissue samples contained a higher viral titer than other tissues. However, the authors did not analyze whether PPgV infections caused histopathological changes in the liver. In this study, we detected the presence of PPgV infection in serum samples from apparently healthy pigs and pigs with clinical symptoms, which is consistent with the findings of previous studies (Baechlein et al. 2016; Kennedy et al. 2019). Thus, an association between PPgV infection and clinical disease is yet to be established. Further studies are needed to elucidate the potential association of PPgV infection with disease and the clinical relevance of PPgV infection in pigs.
Since the initial discovery of PPgV, only four studies have described the molecular epidemiology of PPgV, reporting eight complete polyprotein gene sequences and 22 partial NS3 gene sequences (Baechlein et al. 2016; Chen et al. 2019; Kennedy et al. 2019; Lei et al. 2019; Yang et al. 2018). The NS3 and NS5B genes are two most common criteria to classify the phylogeny of pegiviruses (Smith et al. 2016). In this study, we performed phylogenetic analyses using the complete polyprotein gene sequence, as well as the NS3 and NS5B sequences, to investigate the diversity of the PPgV sequence with sequences reported previously (Fig. 2A, 2B, 2C). The phylogenetic analyses were consistent with each other and the analyses of the sequences revealed that all the sequences could be divided into two large clusters (cluster 1 and cluster 2). In particular, sequences from China, including those obtained in this study and the USA were located in cluster 1 (Fig. 2A, 2B, 2C). The alignment of the complete polyprotein gene sequences showed that the sequences obtained in this study had a higher nucleotide homology (95.0%–96.6%) with the sequences from the USA compared to that of PPgV sequences isolated in Germany (87.4%–87.8%) (Table 3). The phylogenetic analyses and sequence alignment indicated that the PPgV sequences obtained in Guangdong Province have a high genetic similarity to the strains isolated in the USA.
The complete PPgV polyprotein gene sequences isolated from Germany, USA, and China have been uploaded in the GenBank database (Baechlein et al. 2016; Chen et al. 2019; Lei et al. 2019; Yang et al. 2018). Since this is a newly identified virus, the number of sequences available for evolutionary and biological analyses remain insufficient. Guangdong is a large province that is crucial in domestic pig breeding in the Chinese pig industry. The two complete polyprotein sequences and seven NS5B sequences reported in this study could provide crucial information about the genetic diversity, molecular evolution, and geographical distribution of PPgV in China. The data will inform better management of PPgV infections and on the potential association between this emerging virus and infections in domestic animals, such as pigs. Extensive epidemiological studies are needed to monitor the prevalence of PPgV in China and to clarify the pathogenicity of its infection and mode of transmission.
-
This work was supported by Key Laboratory of Zoonosis Prevention and Control of Guangdong Province, the Guangdong Province Pig Industrial System Innovation Team (Grant Number 2018LM1103), the National Key Basic Research Program (Grant Number 2016YFD0500606), the Construction of the First Class Universities (Subject) and Special Development Guidance Special Fund (Grant Number K5174960), and the Fundamental Research Funds for the Central Universities, SCUT (Grant Number D2170320).
-
YX designed the experiments, drafted and revised the manuscript. XW analyzed the data and generated phylogenetic trees. GL, and GS completed the collection of samples. JF, LW, and YC performed the experiments. HY and DH conceived the study and finalized the manuscript. All authors read and approved the final version of the manuscript.
-
The authors declare no conflict of interest.
-
All institutional and National guidelines about the use of animals were followed.


 DownLoad:
DownLoad: