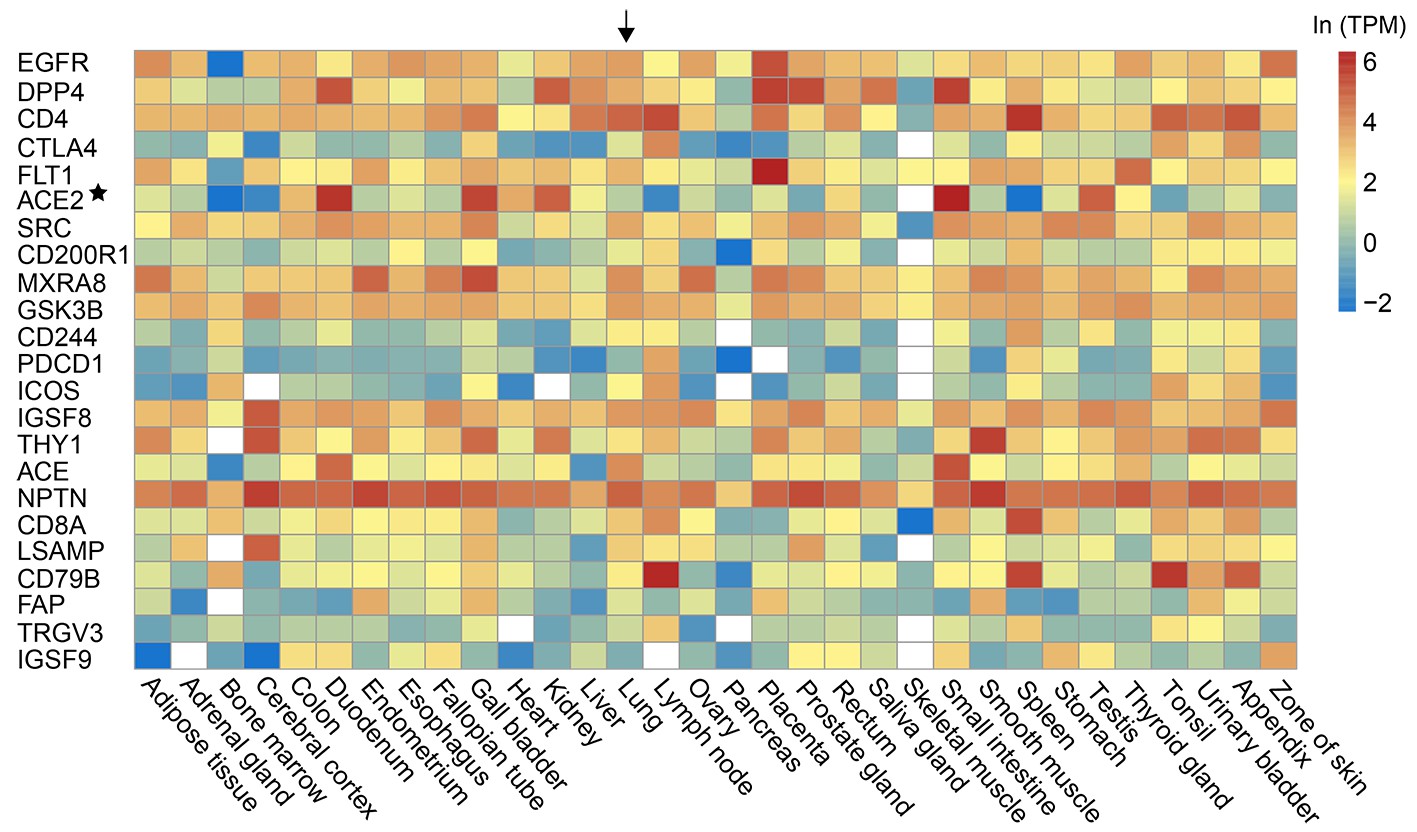
-
Baranowski E, Ruiz-Jarabo CM, Domingo E (2001) Evolution of cell recognition by viruses. Science 292:1102-1105
doi: 10.1126/science.1058613
-
Casasnovas JM (2013) Virus-receptor interactions and receptor-mediated virus entry into host cells. In: Mateu MG (ed) Structure and physics of viruses, vol 68. Springer, Netherlands, pp 441-466
-
Chen C, Liaw A, Breiman L (2004) Using random forest to learn imbalanced data, vol 110. University of California, Berkeley, p 24
-
Csardi G, Nepusz T (2006) The igraph software package for complex network research. Int J Complex Syst 1695:1-9
-
Dimitrov DS (2004) Virus entry: molecular mechanisms and biomedical applications. Nat Rev Microbiol 2:109-122
doi: 10.1038/nrmicro817
-
Free RB, Hazelwood LA, Sibley DR (2009) Identifying novel protein-protein interactions using co-immunoprecipitation and mass spectroscopy. Current Protoc Neurosci 46:5.28.1-5.28.14
-
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150-3152
doi: 10.1093/bioinformatics/bts565
-
Gupta R, Jung E, Brunak S (2004) Prediction of N-glycosylation sites in human proteins. http://www.cbs.dtu.dk/services/NetNGlyc
-
Hoffmann M, Kleine-Weber H, Krueger N, Mueller MA, Drosten C, Pöhlmann S (2020) The novel coronavirus 2019 (2019-nCoV) uses the SARS-coronavirus receptor ACE2 and the cellular protease TMPRSS2 for entry into target cells. BioRxiv. https://doi.org/10.1101/2020.01.31.929042
doi: 10.1101/2020.01.31.929042
-
Lasso G, Mayer SV, Winkelmann ER, Chu T, Elliot O, Patino-Galindo JA, Park K, Rabodan R, Honig B, Shapira SD (2019) A structure-informed Atlas of human-virus interactions. Cell 178(1526-1541):e1516
-
Li F (2015) Receptor recognition mechanisms of coronaviruses: a decade of structural studies. J Virol 89:1954-1964
doi: 10.1128/JVI.02615-14
-
Masson P, Hulo C, De Castro E, Bitter H, Gruenbaum L, Essioux L, Bougueleret L, Xenarios I, Le Mercier P (2012) ViralZone: recent updates to the virus knowledge resource. Nucleic Acids Res 41:D579-D583
doi: 10.1093/nar/gks1220
-
Minor P, Pipkin P, Hockley D, Schild G, Almond J (1984) Monoclonal antibodies which block cellular receptors of poliovirus. Virus Res 1:203-212
doi: 10.1016/0168-1702(84)90039-X
-
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V (2011) Scikit-learn: machine learning in Python. J Mach Learn Res 12:2825-2830
-
Petryszak R, Keays M, Tang YA, Fonseca NA, Barrera E, Burdett T, Füllgrabe A, Fuentes AM-P, Jupp S, Koskinen S (2016) Expression Atlas update-an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res 44:D746-D752
doi: 10.1093/nar/gkv1045
-
Qi F, Qian S, Zhang S, Zhang Z (2020) Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem Biophys Res Commun 526:135-140
doi: 10.1016/j.bbrc.2020.03.044
-
Ryu W-S (2016) Molecular virology of human pathogenic viruses. Academic Press, Amsterdam, pp 247-260
-
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43:D447-D452
doi: 10.1093/nar/gku1003
-
Wang J-h (2002) Protein recognition by cell surface receptors: physiological receptors versus virus interactions. Trends Biochem Sci 27:122-126
doi: 10.1016/S0968-0004(01)02038-2
-
Yan C, Duan G, Wu F-X, Wang J (2019) IILLS: predicting virus-receptor interactions based on similarity and semi-supervised learning. BMC Bioinform 20:651
doi: 10.1186/s12859-019-3278-3
-
Zhang Z, Zhu Z, Chen W, Cai Z, Xu B, Tan Z, Wu A, Ge X, Guo X, Tan Z, Xia Z, Zhu H, Jiang T, Peng Y (2019) Cell membrane proteins with high n-glycosylation, high expression and multiple interaction partners are preferred by mammalian viruses as receptors. Bioinformatics 35:723-728
doi: 10.1093/bioinformatics/bty694
-
Zhang H, Kang Z, Gong H, Xu D, Wang J, Li Z, Cui X, Xiao J, Meng T, Zhou W (2020) The digestive system is a potential route of 2019-nCov infection: a bioinformatics analysis based on single-cell transcriptomes. BioRxiv. https://doi.org/10.1101/2020.01.30.927806
doi: 10.1101/2020.01.30.927806
-
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270-273
doi: 10.1038/s41586-020-2012-7