














HTML
-
Human adenoviruses (HAdVs) are important pathogens and responsible for respiratory infections. HAdVs are widespread in the world; 3.5%-11% of childhood community acquired pneumonias are associated with HAdV infection (Chen et al. 2015; Jain et al. 2015; Liu et al. 2015; Wang et al. 2015). HAdVs are nonenveloped, double-stranded DNA viruses and have been classified into seven species (A-G), including 103 genotypes (http://hadvwg.gmu.edu/). Different genotypes display different tissue tropisms associated with their manifestation of infection. HAdV species B (HAdV-3, 7, 11, 14, 16, 21, 50 and 55), C (HAdV-1, 2, 5 and 6) and E (HAdV-4) are mainly related to respiratory diseases (Knipe and Howley. 2013). In China, HAdV-7 is one of the predominant genotypes, especially in northern China, accounting for 26.9% of all HAdV infections. Many outbreaks and epidemics of HAdV-7 have been reported (Tang et al. 2011; Liu et al. 2015; Duan et al. 2019). Importantly, HAdV-7 infection often causes more severe clinical manifestations and higher mortality than other genotypes; therefore, HAdV-7 has attracted special attention (Kim et al. 2003; Yu et al. 2016).
Vaccines against HAdV-4 and HAdV-7 have been developed only for U.S. military recruits; however, no safe and effective vaccines or drugs for HAdV-7 are available in China (Hoke and Snyder. 2013). Therefore, monitoring the prevalence of HAdV-7 in China and analysing the genetic variation of the HAdV-7 genome, especially in the region of the hexon gene encoding the hypervariable regions 1-7 (Loops 1 and 2), are of great significance to the development of vaccines and drugs against HAdV-7 in China.
In this study, 57 HAdV-7-positive respiratory samples from children with acute respiratory infections were collected in Beijing, Shijiazhuang, Wenzhou and Guangzhou between 2014 and 2018 and selected for hexon, penton base and fiber gene sequencing. Since the obtained sequences of hexon, penton base and fiber have high percent identity, we randomly selected 17 strains from Beijing, Shijiazhuang, Wenzhou and Guangzhou for whole genome sequencing. Phylogenetic, amino acid mutation and recombination analyses were performed on the HAdV-7 sequences. This study was designed to elucidate the genetic characteristics of HAdV-7 strains that are prevalent in China and to provide basic reference data for the prevention, control and vaccine development of HAdV-7.
-
Between 2014 and 2018, 57 HAdV-7-positive specimens were collected from patients in a multicentre study based on a network monitoring viral pathogens of respiratory tract infection in children in four cities (Beijing, Shijiazhuang, Wenzhou and Guangzhou) (Supplementary Table S1). Thirty-three HAdV-7 strains were isolated by cell culture from these 57 HAdV-7-positive specimens. Among them, 17 of 33 strains from different cities were randomly selected for whole genome sequence analysis.
-
DNA was extracted from the specimens or isolates by using a QIAamp MinElute Virus Spin Kit (QIAGEN, Germany) according to the manufacturer's instructions.
-
Polymerase chain reaction (PCR) amplification was conducted using HotStar Taq Plus Master Mix Kits (QIAGEN, Germany) for the hexon, penton base, fiber genes and whole genomic sequence. The primers used are listed in Supplementary Table S2. PCR products were purified by agarose gel electrophoresis, then sequenced using a Sanger primer-walking method by SinoGenoMax Co., Ltd. The sequence data provided at least twofold coverage of the genome. The sequences were assembled and edited with DNASTAR v7.1 (DNASTAR Inc., WI, USA). Sequences of the hexon, penton base and fiber genes were acquired from 57 specimens and strains and 17 whole genomic sequences were obtained (GenBank accession numbers are shown in Supplementary Table S1).
-
Whole genomic reference sequences of HAdV-7 were downloaded from the GenBank database. Reference sequences typed as HAdV-7 only based on the hexon gene without the penton base and fiber genes were excluded, as well as sequences without collection location or date. Finally, 56 whole genomic reference sequences obtained from GenBank were enrolled for phylogenetic analysis. The reference sequences are listed in Supplementary Table S3.
Multiple alignment of the hexon, penton base, fiber genes and the whole genomic sequences were conducted using MAFFT (https://www.ebi.ac.uk/Tools/msa/mafft/) software. Phylogenetic trees were generated by MEGA v6.0 (Sudhir Kumar, Arizona State University, Tempe, AZ, USA) software with the neighbour-joining method and the Kimura 2-parameter model. The reliability of phylogenetic inference was estimated by the bootstrap method with 1000 replicates.
-
BioEdit v7.2.0 (http://www.mbio.ncsu.edu/BioEdit/page2.html) software was used for identity and diversity analysis.
-
Recombination analysis of the whole genomic sequences of the HAdV-7 strains was carried out using Simplot v3.5.1 software with the selected parameters including a window size of 1000, a step size of 200 and Kimura (two parameter) distance correction.
Samples and Strains
DNA Extraction
Hexon, Penton base, Fiber gene and Whole Genomic Sequence analysis
Phylogenetic Analysis
Analysis of Genetic Variation
Recombination Analysis
-
The nucleotide and amino acid identities of the hexon gene obtained in this study were 99.8%-100% and 100%, respectively, while the penton base and fiber genes identities were 99.8%-100% and 99.8%-100% at the nucleotide level and 99.8%-100% and 99.6%-100% at the amino acid level, respectively.
The phylogenetic dendrogram based on the hexon gene sequences formed two clusters. The strains gz07 (HQ659699) and GZ08 (GQ478341) collected from Guangdong in 2007 and 2008, as well as the prototype strain (AY594255) formed cluster 1. The 57 hexon gene sequences in this study were located in cluster 2 with most of the circulating strains worldwide, including 2 vaccine strains (AY495969 and AY594256), from the 1960s to now (Fig. 1). The within-group mean genetic distances were 0.0007 and 0.0004 for cluster 1 and cluster 2, respectively. The mean genetic distance between cluster 1 and cluster 2 was 0.0350.

Figure 1. Phylogenetic analysis of the major capsid genes hexon, penton base and fiber. The phylogenetic trees were generated by using the neighbor-joining method based on the Kimura two-parameter model with 1000 replicates. The red dots indicate the clinical strains obtained in this study. The black dots indicate the vaccine strains. The black triangle indicates HAdV-7 prototype strain (Accession Number is AY594255).
The penton base gene sequences were stratified into 3 branches. Clusters 1 and 2 were represented by the vaccine strains (AY495969 and AY594256) and the prototype strain (AY594255), respectively. Most of the strains circulating worldwide including the 57 sequences from this study formed cluster 3 (Fig. 1). The within-group mean genetic distances were 0.0006, 0.0000 and 0.0003 for cluster 1, cluster 2 and cluster 3, respectively. The mean genetic distances were 0.0170, 0.0170 and 0.0070 between cluster 1 and cluster 2, cluster 1 and cluster 3, cluster 2 and cluster 3 respectively.
The phylogenetic tree of the fiber gene sequences formed 4 clusters. In accordance with the phylogenetic tree based on the hexon gene sequences, the 57 fiber gene sequences in this study were located in the cluster 4 with most of the strains circulating in China, the USA, Russia, as well as two vaccine strains (AY495969 and AY594256). While gz07 (HQ659699), GZ08 (GQ478341) and the prototype strain (AY594255) constituted the cluster 2. These results suggested that gz07 (HQ659699) and GZ08 (GQ478341) maybe the re-emergent of early strains which were not found after 2008. The within-group mean genetic distances were 0.0000 for the cluster 2 and 0.0002 for the cluster 4. Interestingly, strain BJ/CHN/2018 (MH355567) isolated from Beijing in 2018 and strain CL_43 (KF268134) isolated from the USA in 1988 were located in two separate clusters: clusters 1 and 3 (Fig. 1). These strains were distinctly different in the phylogenetic trees based on the hexon and penton base genes, although this result was not supported by high bootstrap values. The mean genetic distances among four clusters were also analysed. The highest between-group mean genetic distance was 0.0180 between cluster 1 and cluster 2 and the lowest was 0.0040 between cluster 3 and cluster 4. These results showed that strains in this study were highly homologous to each other, but were evidently different from gz07 (HQ659699) and GZ08 (GQ478341) which always clustered with the prototype strain (AY594255).
-
Neutralizing antibodies, which are generated after HAdV infection, mainly target the hypervariable regions of hexon (Sumida et al. 2005; Roberts et al. 2006). There are species-specific and type-specific epitopes (ε determinants) on hexon. The hypervariable regions and type-specific domains of hexon are encoded by the loop1 and loop2 sequences. Compared with the HAdV-7 prototype strain (AY594255), the 57 strains in this study had 3 amino acid deletions (R143, A144 and V145) and 10 amino acid substitutions (A139T, E142D, T146N, T147A, N150Y, M157T, T200I, K242T, T250A and D298G) in the loop1 region of the hexon protein (Fig. 2A). Eleven amino acid substitutions were found in the loop2 region (A412G, K413N, T414K, S420P, K421R, N423T, G424A, D429T, N430K, K433T and S434A). Meanwhile, the same mutations were also observed in all strains from China except gz07 (HQ659699) and GZ08 (GQ478341). For gz07 (HQ659699) and GZ08 (GQ478341), which clustered with the prototype strain (AY594255) in the phylogenetic tree of hexon, only one amino acid substitution (Q443L) was found in the loop2 region (Fig. 2B).

Figure 2. Amino acid variation analysis in the loop1 (A) and loop2 (B) of hexon. In the HAdV-7 prototype strain Gomen, the region of loop 1 was 135-304 AA location of hexon and loop 2 was 407-452 AA location of hexon (Pring-Akerblom et al 1995). The fifty-seven sequences of hexon obtained in this study have high identity and the same amino acid variations. Therefore, GZ042 were selected as the representative showing in the figure.
The Arg-Gly-Asp (RGD) motif on the penton base can interact with integrins on the cell surface to accelerate viral infection by promoting the internalization process of the virus (Wickham et al. 1994). Therefore, genetic mutations in the RGD loop region may affect the internalization of HAdV. The amino acid mutation D325N in the RGD loop region was observed in all strains obtained in this study except WZ059 when compared with the prototype strain (AY594255). The same mutation was also found in most circulating strains worldwide. For gz07 (HQ659699) and GZ08 (GQ478341), no amino acid mutation was found in this region (Fig. 3A). The HVR1 region of the penton base is on the surface of the viral capsid. Recombination has been reported in HVR1 region and its vicinity (Madisch et al. 2007), however, all strains from China including 57 sequences of this study are identical to the sequences of the prototype strain (AY594255) in this region, except vaccine strain (AY594256) of China with the substitution I158T (Fig. 3B).

Figure 3. Amino acid variation analysis in the RGD (A) and HVR1 (B) of penton base. In the HAdV-7 prototype strain Gomen, the region of RGD was 299-362 AA location of penton base and HVR1 was 150-169 AA location of penton base (Madisch et al 2007). The fifty-seven sequences of penton base obtained in this study have high identity and the same amino acid variations except WZ059. Therefore, GZ042 were selected to represent all sequences we obtained except WZ059.
-
The nucleotide identity of the 17 complete genomes obtained in this study was 99.7%-99.9%. Phylogenetic analysis of the whole genome sequences showed that all HAdV-7 strains could be stratified into 3 clusters which were consistent with the phylogenetic tree of the penton base gene. The cluster 1 was composed of prototype strain (AY594255), gz07 (HQ659699) and GZ08 (GQ478341). The two vaccine strains (AY495969 and AY594256) were located in the cluster 2, as well as the USA strains 55, 142 (MH910669, in 1960s) and CL_43 (KF268134, in 1988). The 17 whole genome sequences were located in the cluster 3 with the rest of strains circulating in China, the USA and Russia from 1988 to 2018, independent of the prototype strain (AY594255) or vaccine strains (AY495969 and AY594256). Cluster 3 could be further divided into two branches. Three strains collected from USA before 2000 constituted one branch with a bootstrap value of 99%. All strains collected after 2000 formed the other branch besides the strain ak39_AdV7d2 (JX423387) collected from USA in 1997. Whereas, the strains from different countries had no obvious geographical aggregation. The within group mean genetic distances were 0.0002, 0.0013 and 0.0006 for cluster 1, cluster 2 and cluster 3. The between group mean genetic distances were 0.018, 0.019 and 0.006 between cluster 1 and cluster 2, cluster 1 and cluster 3, cluster 2 and cluster 3, respectively (Fig. 4). These results showed that strains in this study have no significant differences from those collected in China, the USA and Russia from 1988 to 2018.
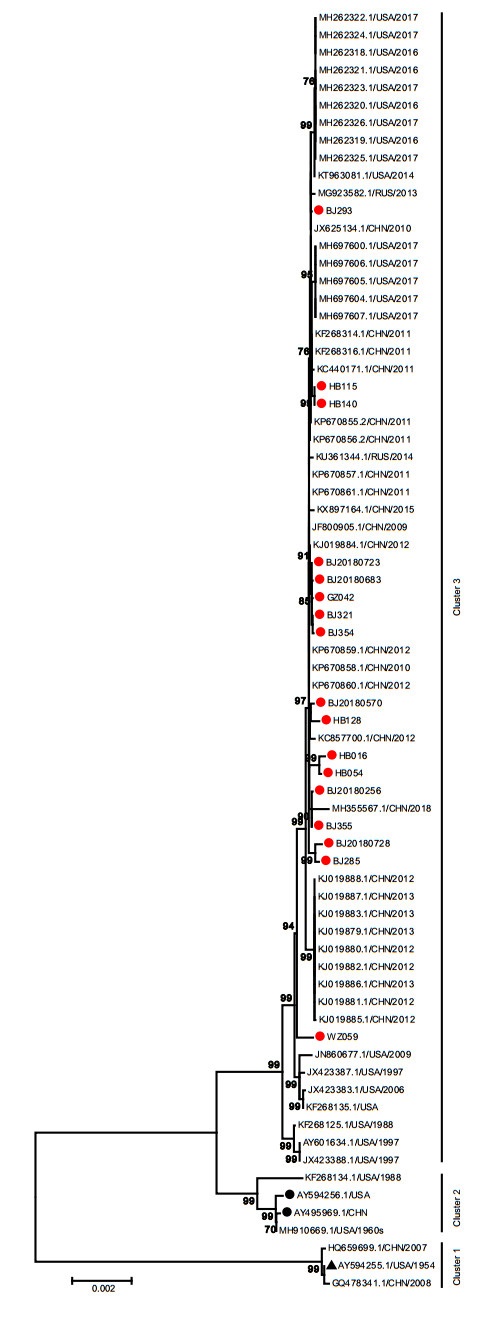
Figure 4. Phylogenetic analysis of complete genome of HAdV-7. The phylogenetic tree was generated by using the neighbor-joining method based on the Kimura two-parameter model with 1000 replicates. The red dots indicate the clinical strains obtained in this study. The black dots indicate the vaccine strains. The black triangle indicates HAdV-7 prototype strain (Accession Number is AY594255).
-
The early genes of HAdV include the E1, E2, E3 and E4 genes, which are important for viral transcription, replication and interference with host immunity (Tauber and Dobner. 2001; Zeng and Carlin. 2019). We found that the phylogenetic dendrograms based on the E1, E2A, E2B and E3 sequences were consistent with the tree based on hexon gene sequences and 2 clusters were formed. The prototype strain (AY594255) was together with gz07 (HQ659699) and GZ08 (GQ478341) in cluster 1. However, the 17 early gene sequences in this study got together with most circulating strains worldwide, including 2 vaccine strains (AY495969 and AY594256) in cluster 2 (Fig. 5).

Figure 5. Phylogenetic analysis of E1, E2A, E2B, E3 gene. The phylogenetic trees were generated by using the neighbor-joining method based on the Kimura two-parameter model with 1000 replicates. The red dots indicate the clinical strains obtained in this study. The black dots indicate the vaccine strains. The black triangle indicates HAdV-7 prototype strain (Accession Number is AY594255).
The E4 gene phylogenetic tree revealed 4 clusters. The E4 gene sequences in this study were together with most circulating strains worldwide isolated from 1988 to present in cluster 4. Consistent with the phylogenetic tree of the E1, E2 and E3 genes, the prototype strain (AY594255), gz07 (HQ659699) and GZ08 (GQ478341) formed cluster 1. The 2 vaccine strains (AY495969 and AY594256) were located in cluster 2 with strain 55, 142 (MH910669). Unexpectedly, the strain CL_43 (KF268134) was the only member of cluster 3 (Fig. 6).
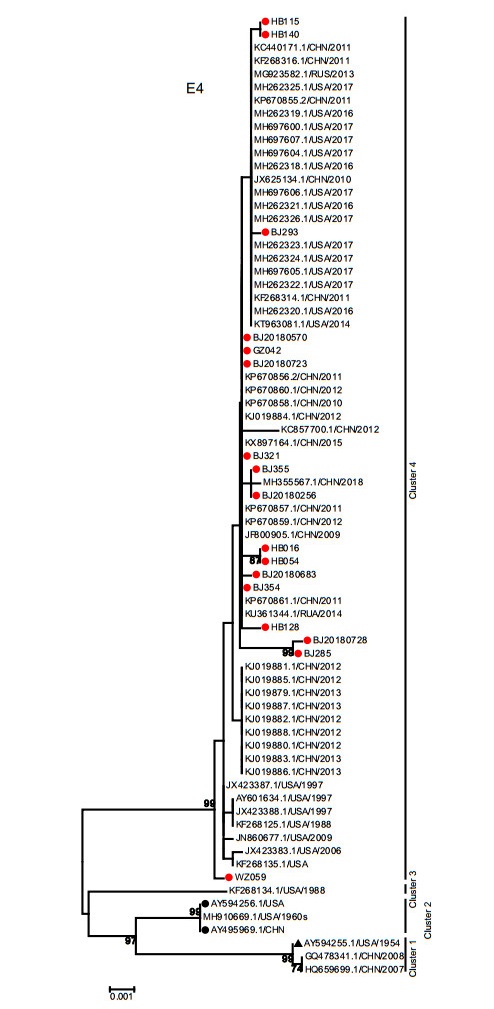
Figure 6. Phylogenetic analysis of E4 gene. The phylogenetic trees were generated by using the neighbor-joining method based on the Kimura two-parameter model with 1000 replicates. The red dots indicate the clinical strains obtained in this study. The black dots indicate the vaccine strains. The black triangle indicates HAdV-7 prototype strain (Accession Number is AY594255).
Phylogenetic Analysis of the Hexon, Penton Base and Fiber Genes
Amino Acid Variation Analysis
Phylogenetic Analysis of the Complete Genome
Phylogenetic Analysis of Early Genes
-
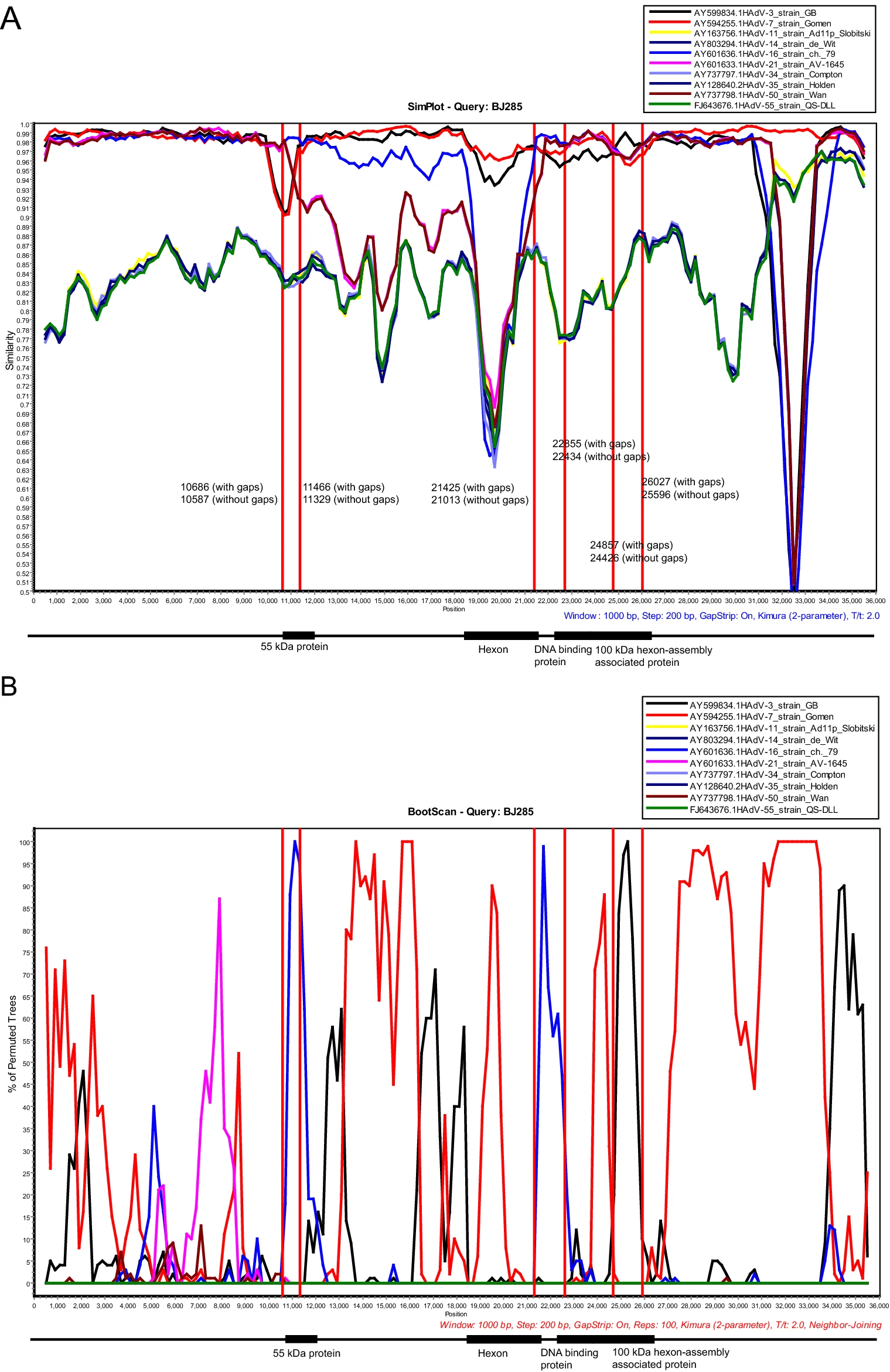
Recombination is an important mechanism for HAdV evolution. The recombination analysis of these 17 complete genomes showed that 3 gene fragments were recombined from HAdV-16 and HAdV-3. Partial region of 55 kDa protein gene of the HAdV-7 strains was recombined from HAdV-16, while partial region of 100 kDa hexon-assembly associated protein gene of the HAdV-7 strains was recombined from HAdV-3. The remaining recombinant gene fragment was derived from HAdV-16 with beginning breakpoint located around 21, 013 (without gaps) of HAdV-7 (BJ285), within the gene coding for hexon. And the ending breakpoint located around 22, 434 (without gaps) of HAdV-7 (BJ285), within the gene coding for DNA binding protein (Fig. 7 and Supplementary Fig. S1). Recombination analysis on the strain S 1058 (MH910662) isolated in 1955, the earliest strain among the reference sequences except the prototype strain (AY594255), also indicated a similar recombination event. Therefore, we deduced that a recombination event occurred as early as 1955 (Supplementary Fig. S2). The results coincided with phylogenetic analysis of the three recombinant regions, which indicated that both of recombinant region 1 (10, 587-11, 329 nucleotide gene location of BJ285, partial region of 55 kDa protein gene) and recombinant region 2 (21, 013-22, 434 nucleotide gene location of BJ285, partial regions of hexon and DNA binding protein gene) clustered with HAdV-16 prototype (Supplementary Fig. S3A, S3B), but the recombinant region 3 (24, 426-25, 596 nucleotide gene location of BJ285, partial region of 100 kDa hexon-assembly associated protein) clustered with HAdV-3 prototype (Supplementary Fig. S3C).
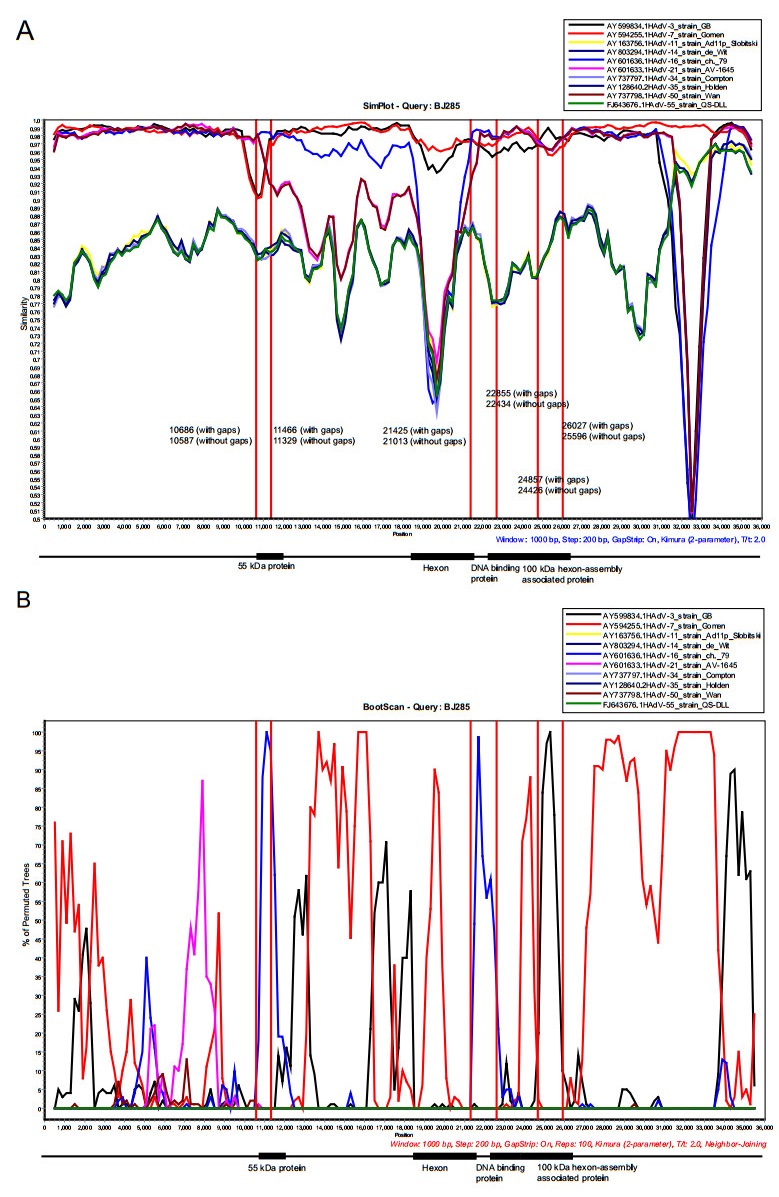
Figure 7. Genome recombination analysis. (A) Simplot and (B) Bootscan analysis of the whole genomes of strain BJ285 compared with other species B adenoviruses. Recombination analysis was performed by using Simplot with the following input: window size (1000 nucleotides [nt]), step size (200 nt), distance model (Kimura) and tree model (neighbor-joining). The GenBank accession numbers of prototype strains of each HAdV are as follows: HAdV-3, AY599834; HAdV-7, AY594255; HAdV-11, AY163756; HAdV-14, AY803294; HAdV-16, AY60 1636; HAdV-21, AY60 1633; HAdV-34, AY737797; HAdV-35, AY128640; HAdV-50, AY737798; HAdV-55, FJ643676. The genome sequences of the 17 strains obtained in this study have high percent identity and the results of recombination analysis were consistent. Therefore, BJ285 was selected as the representative to display the results of recombination analysis. 55 kDa protein, 10, 833-12, 002 nucleotide gene location of BJ285, without gaps; 10, 970-12, 145 nucleotide gene location, with gaps. Hexon protein, 18, 397-21, 201 nucleotide gene location of BJ285, without gaps; 18, 675-21, 613 nucleotide gene location, with gaps. DNA binding protein, 21, 956-23, 509 nucleotide gene location of BJ285, without gaps; 22, 377-23, 934 nucleotide gene location, with gaps. 100 kDa hexon-assembly associated protein, 23, 540-26, 029 nucleotide gene location of BJ285, without gaps; 23, 965-26, 460 nucleotide gene location, with gaps.
-
This study performed phylogenetic analyses on the hexon, penton base and fiber sequences of 57 clinical strains and the whole genome sequences of 17 HAdV-7 isolates collected from four cities in northern and southern China from 2014 to 2018. The phylogenetic dendrogram based on the whole genome and penton base sequences formed three clusters. The phylogenetic tree of the hexon, E1, E2A, E2B and E3 sequences formed two identical clusters. However, the fiber and E4 gene sequences were stratified into four branches. We found that the sequences of the HAdV-7 strains obtained in this study were always located in the clusters with most strains collected from different areas around the world from the 1980s to 2017 in all phylogenetic trees. Strains gz07 (HQ659699), GZ08 (GQ478341) and prototype strain (AY594255) were clustered into the other cluster at all times. We also observed that the mean genetic distances within those clusters were very low (< 0.001). The strains we obtained had high percent identity and high homology with most other strains from different regions and countries. Therefore, the genomes of the HAdV-7 strains currently circulating in China are relatively stable across time and geographic space. Yu et al. (2016) also reported that genomes of two isolates(2011, 2012) in southern China had 100% identical with an earlier strain (2009) in northern China, which suggested the genomic conservation and stability of HAdV-7.
Phylogenetic analysis of the hexon sequences showed that the prototype strain, the vaccine strains and the strains obtained in this study were located in two different clusters. This finding was consistent with the results of phylogenetic trees based on loop1 and loop2 of hexon of HAdV-7 strains isolated from 1954-1987 reported in previous studies (Li and Wadell 1999). According to the analysis of the migration degree of pairwise comigrating restriction fragments of HAdV DNA, HAdV-7 was divided into three clusters in previous studies (Schmitz et al. 1983). In our study, the whole genome sequences of HAdV-7 were also stratified into three branches and the prototype HAdV-7p was located in a single cluster as well. These results indicate that HAdV-7 genomes are relatively conserved with limited mutations.
More than 20 genomic variants of HAdV-7 were identified by restriction enzyme analysis prior to the widespread implementation of viral whole-genome sequencing (Li and Wadell 1986; Larranaga et al. 2000; Ikeda et al. 2003; Kim et al. 2003). In the phylogenetic tree of the whole genome sequences, only the prototype strain initially typed as HAdV-7p and the vaccine strain typed as HAdV-7a were located in independent clusters. Other genomic variants, such as HAdV-7b and HAdV-7d, were all clustered in the same cluster with 17 strains in this study (Fig. 4 and Supplementary Table S3). Although restriction endonuclease analysis with the advantages of low-price and speediness still is a useful technique for characterizing viral pathogens (Zhang et al. 2016), the restriction enzyme used in the earlier typing of HAdV-7 can only cut certain sequences (Li and Wadell 1986), this method cannot fully reflect the molecular evolution characteristics of the whole genome. Therefore, the most reliable way to type HAdV when a large number of recombinant strains are found is through whole genome sequencing analysis (http://hadvwg.gmu.edu/).
By comparison with the prototype strain, amino acid mutations were observed at some antigen neutralization sites in the hexon gene of the HAdV-7 strains obtained in this study. These changes included the replacement of hydrophilic/hydrophobic amino acids (A139T, T147A, M157T, T200I, T250A and S434A), substitution of a heterocyclic amino acid (S420P) and deletion of other amino acids (R143, A144 and V145). Similar amino acid substitutions and deletions have been reported in previous studies (Crawford-Miksza et al. 1999; Li and Wadell 1999). Li et al. showed that genomic variants of HAdV-7 (HAdV-7b, 7c, 7d, 7g and 7h) had consistent amino acid mutations in the loop1 and loop2 of hexon (Li and Wadell 1999). Amino acid variation analysis of reference strains from other countries was also performed. The reference strains from other countries had the same amino acid mutations as Chinese strains, however, Q443L and S432Y were only found in foreign reference strains. These mutations might affect the secondary structure of proteins and are more likely to alter the appearance and properties of antigens (Li and Wadell 1999). In a vaccine study, cross-neutralization tests revealed that the neutralization titer of rabbit antiserum to HAdV-7a standardized by the National Institutes of Health was the same between HAdV-7a strains and the vaccine strain and was eight times to that of the HAdV-7 prototype strain (Crawford-Miksza et al. 1999). It is possible that the amino acid substitutions and deletions resulted in the difference in the antigenic characteristics between the vaccine strain and the prototype strain. The loop1 and loop2 sequences of hexon were basically identical between the strains in this study and the HAdV-7 vaccine strains. Further studies are needed to evaluate the impact of these amino acids on antigenicity.
Recombination is an important mechanism for the evolution of HAdV. Most genotypes of species HAdV-D show evidence of recombination events (Houldcroft et al. 2018). HAdV-89 of species C is also derived from recombination (Dhingra et al. 2019). Zhao et al. (Zhao et al. 2014) reported that the 55 kDa protein gene of HAdV-7d was recombined from HAdV-16. Similar to Zhao's results, we also found the recombination event of HAdV-7. Recombination analysis of 17 HAdV-7 strains in this study demonstrated that partial regions of 55 kDa protein gene and 100 kDa hexon-assembly associated protein gene of these strains were recombined from HAdV-16 and HAdV-3, respectively. The 55 kDa protein is a DNA-binding protein expressed in the early and late stages of infection and plays multiple roles in the HAdV life cycle. The 55 kDa protein can form a complex with the IVa2 protein and is absolutely necessary for DNA packaging (Gustin et al. 1996; Chahal and Flint. 2012). The 100 kDa hexon-assembly associated protein is responsible for hexon assembly and necessary for efficient translation of late viral mRNA (Oosterom-Dragon and Ginsberg. 1981; Hayes et al. 1990). At present, the impact of these recombination events on the virulence and pathogenicity of HAdV-7 are not clear and further researches are needed.
There are also some limitations of this study due to the amino acid variation analysis only performed on hexon and penton that lack of the identification of genomic variants. In addition, the coverage in the whole genome sequencing approach was relatively low.
In conclusion, although our study indicated that the HAdV-7 strains circulating in China were relatively stable and had high percent identities of nucleotide and amino acid, some amino acid variations were found at antigenic sites in loop1 and loop2 of the hexon gene and RGD loop of the penton base gene. Further studies are needed to evaluate the impact of these amino acid mutations on antigenicity. The 55 kDa protein and 100 kDa hexon-assembly associated protein genes of HAdV-7 clinical strains were recombined from HAdV-16 and HAdV-3, respectively and these events occurred as early as 1955. Our study comprehensively described the molecular evolution characteristics of HAdV-7 circulating in China and provided basic reference data for the development of anti-HAdV-7 vaccines and drugs.
-
This work was funded by the Key Technology R&D Program of China (grant numbers2017ZX10103004-004, 2017ZX10104001-005-010), National Natural Science Foundation of China (grant number 82072266), CAMS Innovation Fund for Medical Sciences (CIFMS), (grant number 2019-I2M-5-026).
-
YD, XC and ZX conceived and designed the experiments. YD, CL, LD, SA, WW, ZM and BX performed the experiments. YD, XC, YZ and ZX analyzed the data. YD, XC and ZX wrote the manuscript and prepared the Figures. YD, XC, LX and ZX checked and finalized the manuscript. ZX and XC provided resources. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest.
-
This study was approved by the Medical Ethics Committee of the Beijing Children's Hospital affiliated to Capital Medical University and informed consent was obtained. All procedures were conducted according to the guidelines (ethics number 2017-k-15).


 DownLoad:
DownLoad: