-
 Due to our negligence, the original version of this article, published online on 17 June 2022, contained a mistake in Table 2. The positive animal number for unclassified goats/sheep in the fourth line should be 44. The seropositive rate "3.1%" is correct thus remains unchanged. The corrected Table 2 is given below. We apologize for our oversight when preparing the table and state that this does not change the scientific conclusions of the article in any way.
Due to our negligence, the original version of this article, published online on 17 June 2022, contained a mistake in Table 2. The positive animal number for unclassified goats/sheep in the fourth line should be 44. The seropositive rate "3.1%" is correct thus remains unchanged. The corrected Table 2 is given below. We apologize for our oversight when preparing the table and state that this does not change the scientific conclusions of the article in any way. -
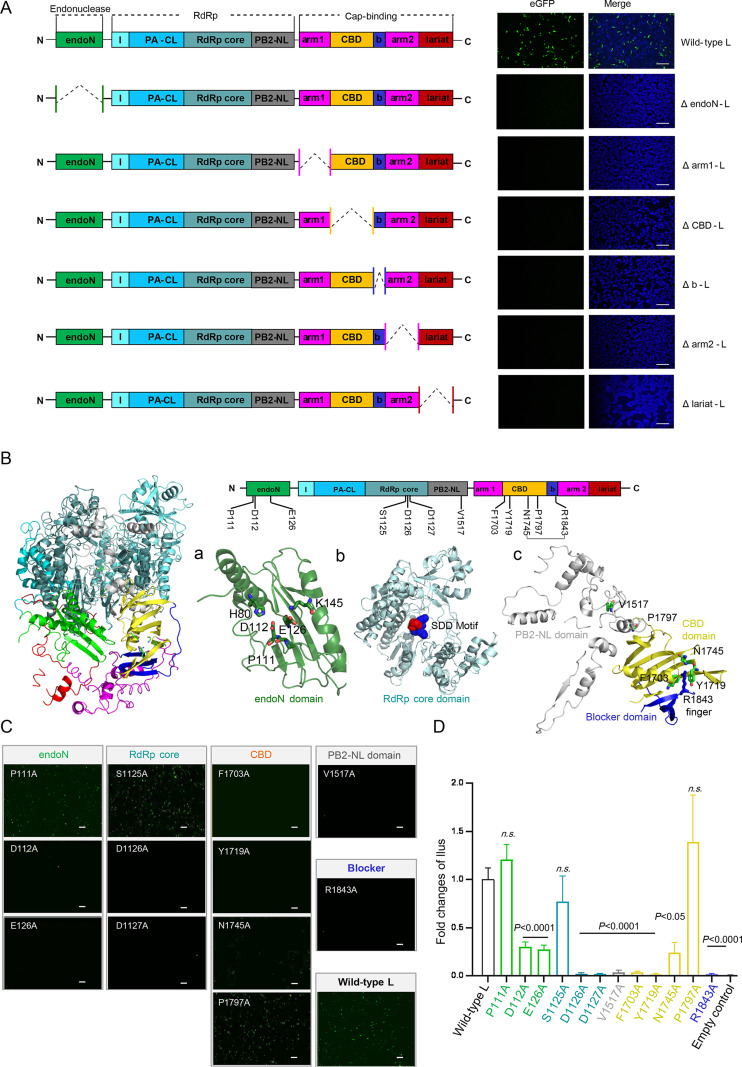
Roles of the functional domains and conserved residues of the severe fever with thrombocytopenia syndrome virus L protein provide insights into the viral RNA transcription/replication mechanism
2022, 37(6): 946 doi: 10.1016/j.virs.2022.08.009
Received: 17 June 2022 Accepted: 26 August 2022Highlights
1. All the domains of SFTSV-L protein were required for its function in viral RNA replication/transcription.
2. The influence of twelve conserved residues speculated basing on the atomic model of SFTSV-L were evaluated using mini-genome system, and nine of which showed significant roles in the function of L protein.
3. Nine key residues of SFTSV-L were strictly conserved among emerging related phleboviruses, suggesting L protein may be a promising broad spectrum drug target of phleboviruses. -
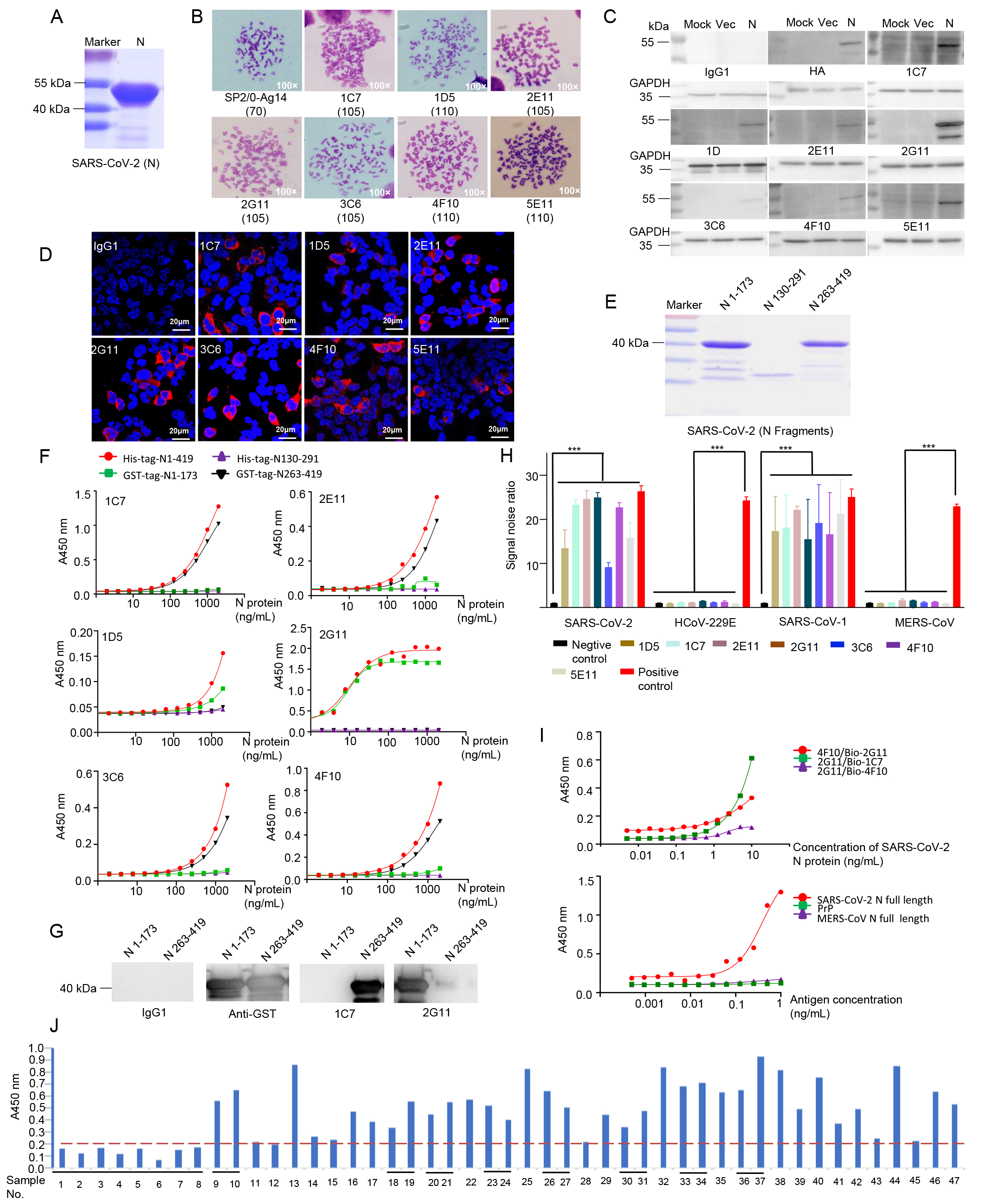
A pair of SARS-CoV-2 nucleocapsid protein monoclonal antibodies shows high specificity and sensitivity for diagnosis
2022, 37(6): 942 doi: 10.1016/j.virs.2022.10.003
Received: 14 February 2022 Accepted: 17 October 2022Highlights
1. Seven monoclonal antibodies (mAbs) against SARS-CoV-2 nucleocapsid protein are produced, which can be applied in ELISA, Western blotting, and immunofluorescence staining.
2. A pair of mAbs, 2G11/bio-1C7, can detect SARS-CoV-2 nucleocapsid protein as low as 15 pg/well in the double sandwich ELISA.
3. The mAb, 2G11, shows 97.4% sensitivity and 100% specificity for diagnosing the human blood samples. -

Structures of SARS-CoV-2 spike protein alert noteworthy sites for the potential approaching variants
2022, 37(6): 938 doi: 10.1016/j.virs.2022.11.003
Received: 30 June 2022 Accepted: 04 November 2022Highlights
1 Deletion of residues 156–157 warps the neighboring beta-sheet and leads NTD and RBD to shift.
2 T859N stabilizes the packing of the 630 loop motif to make RBD standing transition more difficult.
3 The overall structures of the closed state S complex from different variants resemble each other.
4 Mutations in FPPR may affect the overall structure of the trimeric spike protein. -

SARS-CoV-2 infection induces testicular injury in Rhesus macaque
2022, 37(6): 934 doi: 10.1016/j.virs.2022.10.008
Received: 24 May 2022 Accepted: 21 October 2022Highlights
1 The infection of SARS-CoV-2 lead to varying degrees of testicular pathological damage.
2 The NP antigen of SARS-CoV-2 was not found in male reproductive system of rhesus macaque.
3 Infection-associated inflammatory insult and sex hormone fluctuations may account for the testicular pathophysiology. -
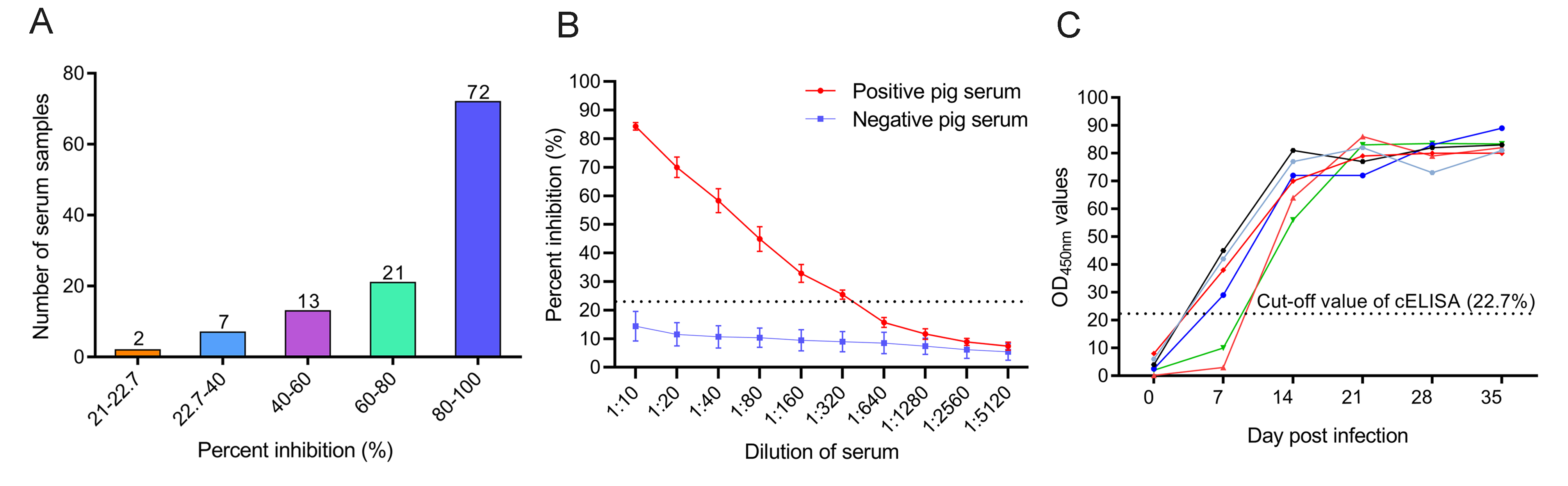
A simple nanobody-based competitive ELISA to detect antibodies against African swine fever virus
2022, 37(6): 922 doi: 10.1016/j.virs.2022.09.004
Received: 04 May 2022 Accepted: 17 July 2022African swine fever virus (ASFV) infection is a big threat to the global pig industry. Because there is no effective vaccine, rapid, low-cost, and simple diagnosis methods are necessary to detect the ASFV infection in pig herds. Nanobodies, with advantages of small molecular weight and easy genetic engineering, have been universally used as reagents for developing diagnostic kits. In this study, the recombinant ASFV-p30 was expressed and served as an antigen to immunize the Bactrian camel. Then, seven nanobodies against ASFV-p30 were screened using phage display technique. Subsequently, the seven nanobodies fused horseradish peroxidase (nanobody-HRP) were secretory expressed and one fusion protein ASFV-p30-Nb75-HRP was selected with the highest sensitivity in blocking ELISA. Using the ASFV-p30-Nb75-HRP fusion protein as a probe, a competitive ELISA (cELISA) was developed for detecting anti-ASFV antibodies in pig sera. The cut-off value of cELISA was determined to be 22.7% by testing 360 negative pig sera. The detection limit of the cELISA for positive pig sera was 1:320, and there was no cross-reaction with anti-other swine virus antibodies. The comparative assay showed that the agreement of the cELISA with a commercial ELISA kit was 100%. More importantly, the developed cELISA showed low cost and easy production as a commercial kit candidate. Collectively, a simple nanobody-based cELISA for detecting antibodies against ASFV is developed and it provides a new method for monitoring ASFV infection in the pig herds. -
Cyclophilin A binds to AKT1 and facilitates the tumorigenicity of Epstein-Barr virus by mediating the activation of AKT/mTOR/NF-κB positive feedback loop
2022, 37(6): 913 doi: 10.1016/j.virs.2022.09.001
Received: 17 January 2022 Accepted: 31 August 2022The AKT/mTOR and NF-κB signalings are crucial pathways activated in cancers including nasopharyngeal carcinoma (NPC), which is prevalent in southern China and closely related to Epstein-Barr virus (EBV) infection. How these master pathways are persistently activated in EBV-associated NPC remains to be investigated. Here we demonstrated that EBV-encoded latent membrane protein 1 (LMP1) promoted cyclophilin A (CYPA) expression through the activation of NF-κB. The depletion of CYPA suppressed cell proliferation and facilitated apoptosis. CYPA was able to bind to AKT1, thus activating AKT/mTOR/NF-κB signaling cascade. Moreover, the use of mTOR inhibitor, rapamycin, subverted the activation of the positive feedback loop, NF-κB/CYPA/AKT/mTOR. It is reasonable that LMP1 expression derived from initial viral infection is enough to assure the constant potentiation of AKT/mTOR and NF-κB signalings. This may partly explain the fact that EBV serves as a tumor-promoting factor with minimal expression of the viral oncoprotein LMP1 in malignancies. Our findings provide new insight into the understanding of causative role of EBV in tumorigenicity during latent infection. -

A novel IFNbeta-induced long non-coding RNA ZAP-IT1 interrupts Zika virus replication in A549 cells
2022, 37(6): 904 doi: 10.1016/j.virs.2022.08.003
Received: 29 December 2021 Accepted: 09 August 2022Zika virus (ZIKV) infection can cause severe neurological diseases including neonatal microcephaly and Guillain-Barre syndrome. Long noncoding RNAs (lncRNAs) are the by-products of the transcription process, which are considered to affect viral infection. However, it remains largely unexplored whether host lncRNAs play a role in ZIKV infection. Here, we identified a group of human lncRNAs that were up-regulated upon ZIKV infection and were dependent on the type I interferon (IFN) signaling. Overexpression of lncRNA ZAP-IT1 leads to an impairment of ZIKV infection. Correspondently, deficiency of ZAP-IT1 led to an enhancement of ZIKV infection. We further confirmed that ZAP-IT1, an intronic lncRNA with total 551 nt in length, is mainly located in the nuclear upon ZIKV infection. Knockout of ZAP-IT1 also led to the increase of dengue virus (DENV), Japanese encephalitis virus (JEV), or vesicular stomatitis virus (VSV) infection. Mechanically, we found that the antiviral effect of ZAP-IT1 was independent of the type I IFN signaling pathway. Therefore, our data unveiled that host lncRNA ZAP-IT1 induced by the type I IFN signaling, showed robust restriction on ZIKV infection, and even on DENV, JEV, and VSV infection, which may benefit the development of antiviral therapeutics. -
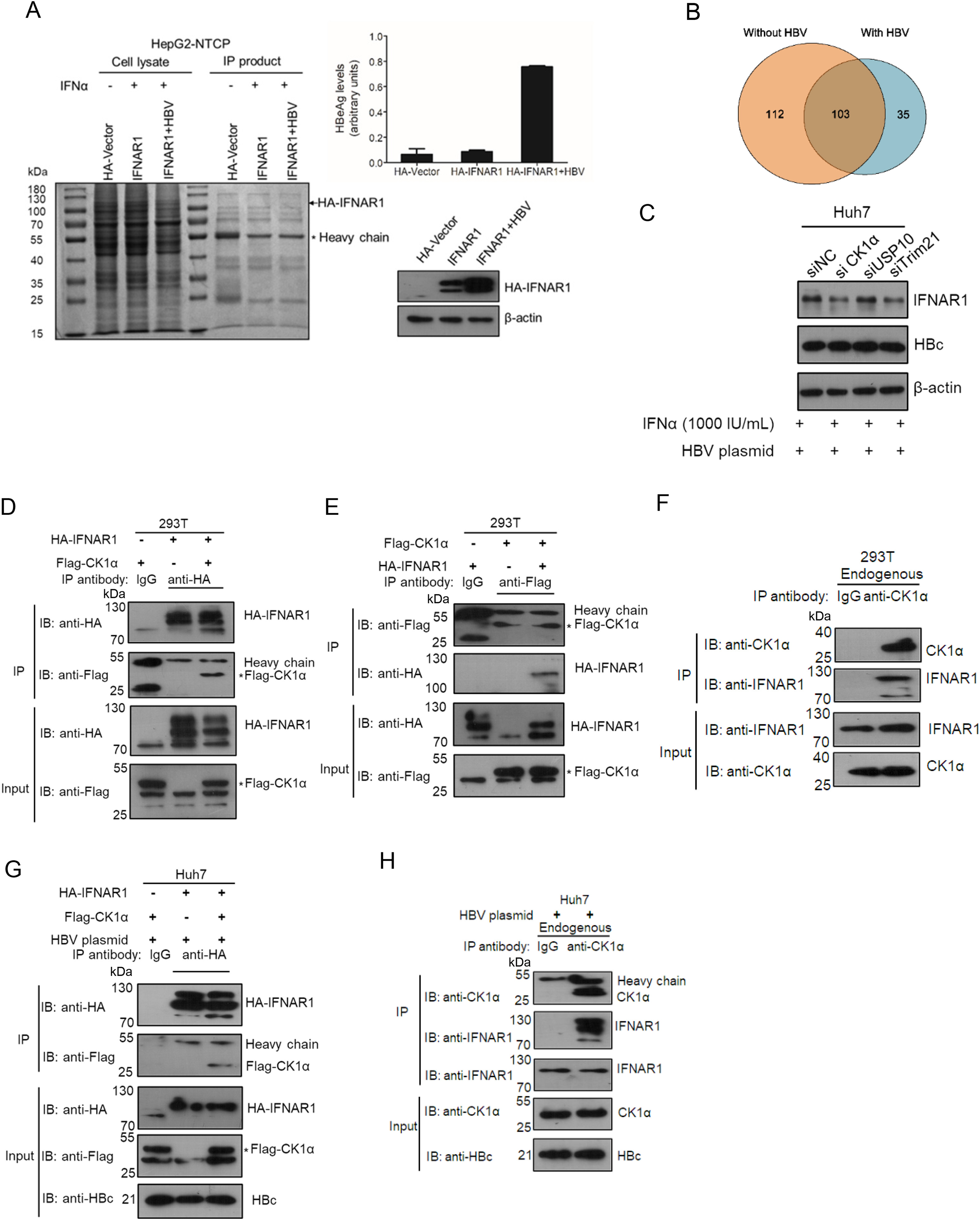
CK1α upregulates the IFNAR1 expression to prompt the anti-HBV effect of type I IFN in hepatoma carcinoma cells
2022, 37(6): 894 doi: 10.1016/j.virs.2022.08.004
Received: 27 January 2022 Accepted: 08 August 2022Casein kinase 1α (CK1α) mediates the phosphorylation and degradation of interferon-α/β receptor 1 (IFNAR1) in response to viral infection. However, how CK1α regulates hepatitis B virus (HBV) replication and the anti-HBV effects of IFN-α are less reported. Here we show that CK1α can interact with IFNAR1 in hepatoma carcinoma cells and increased the abundance of IFNAR1 by reducing the ubiquitination levels in the presence of HBV. Furthermore, CK1α promotes the IFN-α triggered JAK-STAT signaling pathway and consequently enhances the antiviral effects of IFN-α against HBV replication. Our results collectively provide evidence that CK1α positively regulates the anti-HBV activity of IFN-α in hepatoma carcinoma cells, which would be a promising therapeutic target to improve the effectiveness of IFN-α therapy to cure CHB. -
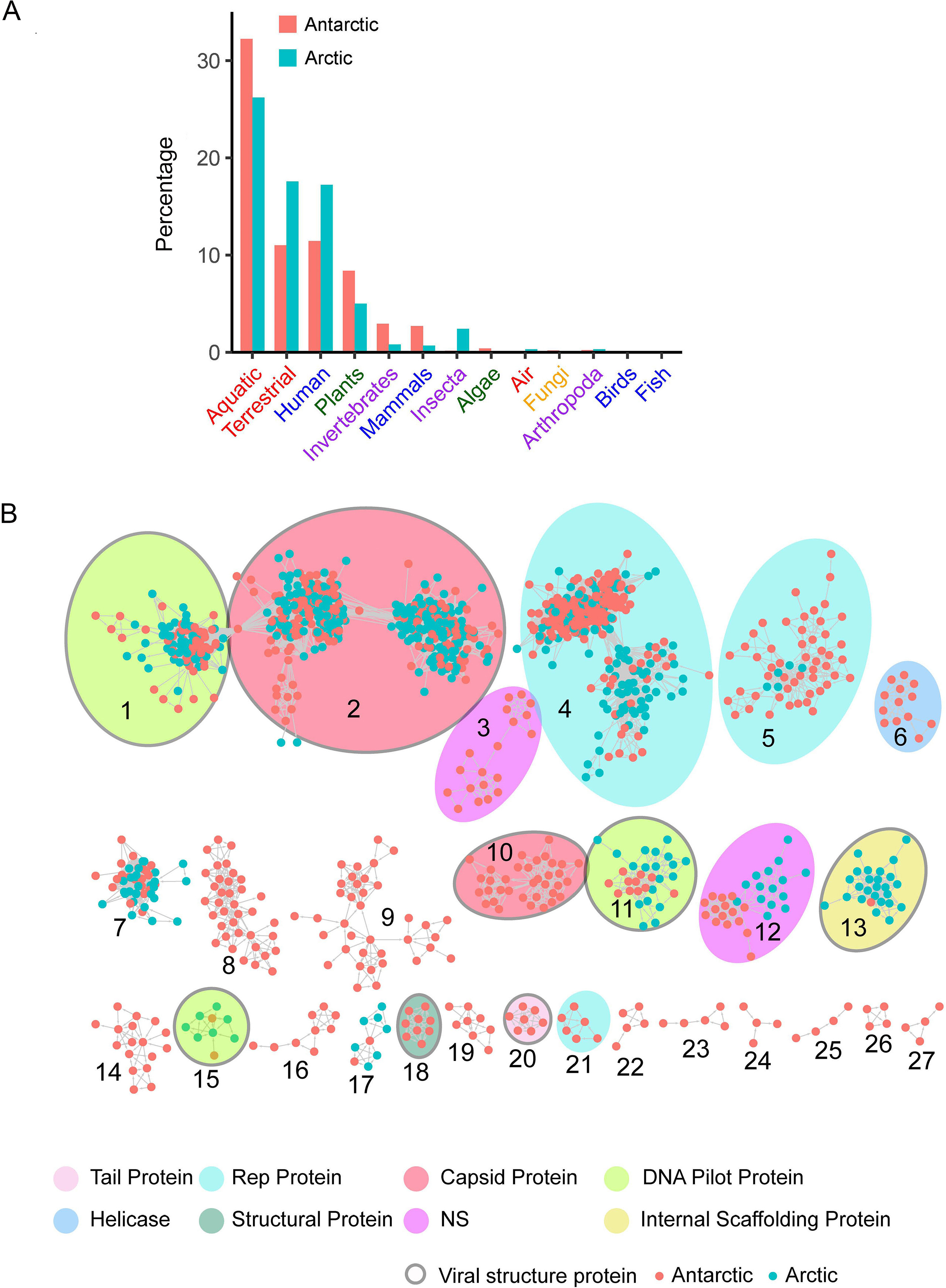
Diverse viromes in polar regions: A retrospective study of metagenomic data from Antarctic animal feces and Arctic frozen soil in 2012–2014
2022, 37(6): 883 doi: 10.1016/j.virs.2022.08.006
Received: 29 April 2022 Accepted: 17 August 2022Antarctica and the Arctic are the coldest places, containing a high diversity of microorganisms, including viruses, which are important components of polar ecosystems. However, owing to the difficulties in obtaining access to animal and environmental samples, the current knowledge of viromes in polar regions is still limited. To better understand polar viromes, this study performed a retrospective analysis using metagenomic sequencing data of animal feces from Antarctica and frozen soil from the Arctic collected during 2012–2014. The results reveal diverse communities of DNA and RNA viruses from at least 23 families from Antarctic animal feces and 16 families from Arctic soils. Although the viral communities from Antarctica and the Arctic show a large diversity, they have genetic similarities with known viruses from different ecosystems and organisms with similar viral proteins. Phylogenetic analysis of Microviridae, Parvoviridae, and Larvidaviridae was further performed, and complete genomic sequences of two novel circular replication-associated protein (rep)-encoding single-stranded (CRESS) DNA viruses closely related to Circoviridae were identified. These results reveal the high diversity, complexity, and novelty of viral communities from polar regions, and suggested the genetic similarity and functional correlations of viromes between the Antarctica and Arctic. Variations in viral families in Arctic soils, Arctic freshwater, and Antarctic soils are discussed. These findings improve our understanding of polar viromes and suggest the importance of performing follow-up in-depth investigations of animal and environmental samples from Antarctica and the Arctic, which would reveal the substantial role of these viruses in the global viral community. -
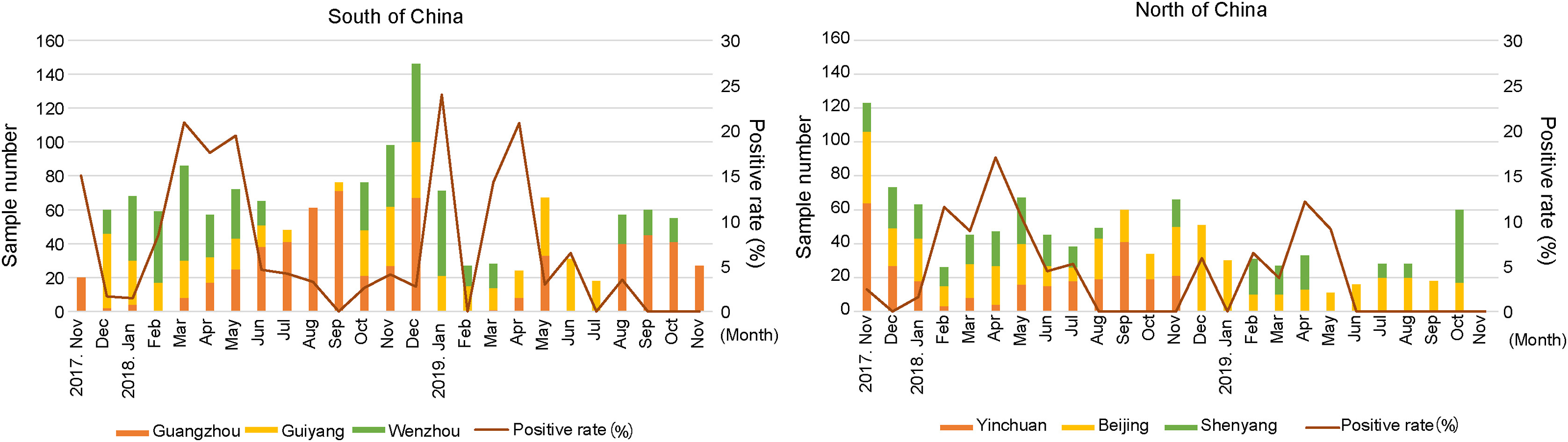
Clinical characteristics and molecular epidemiology of human metapneumovirus in children with acute lower respiratory tract infections in China, 2017 to 2019: A multicentre prospective observational study
2022, 37(6): 874 doi: 10.1016/j.virs.2022.08.007
Received: 06 April 2022 Accepted: 21 June 2022Human metapneumovirus (HMPV) infection is one of the leading causes of hospitalization in young children with acute respiratory illness. In this study, we prospectively collected respiratory tract samples from children who were hospitalized with acute lower respiratory tract infection in six hospitals in China from 2017 to 2019. HMPV was detected in 145 out of 2733 samples (5.3%) from the hospitalized children. The majority of HMPV-positive children were under the age of two (67.6%), with a median age of one year. HMPV can independently cause acute lower respiratory tract infection in young children, while all patients showed mild clinical symptoms. Of all the co-infected patients, HMPV was most commonly detected with enterovirus (EV) or rhinovirus (RhV) (38.0%, followed by respiratory syncytial virus (RSV) (32.0%). The highest detection rate occurred from March to May in both northern and southern China. Out of 145 HMPV positive samples, 48 were successfully typed, of which 36 strains were subgrouped into subtypes A2c (75%), eight strains were included in subtype B1 (16.7%), and four strains were included in subtype B2 (8.3%). Moreover, 16 A2c strains contained 111-nucleotide duplications in the G gene. Twenty-seven complete HMPV genomes were successfully obtained, and 25 (92.6%) strains belonged to subtype A2c, whereas one strain was included in subgroup B1 and another was included in subgroup B2. A total of 277 mutations were observed in the complete genomes of 25 A2c strains. All results presented here improve our understanding of clinical characteristics and molecular epidemiology of HMPV infection in children. -
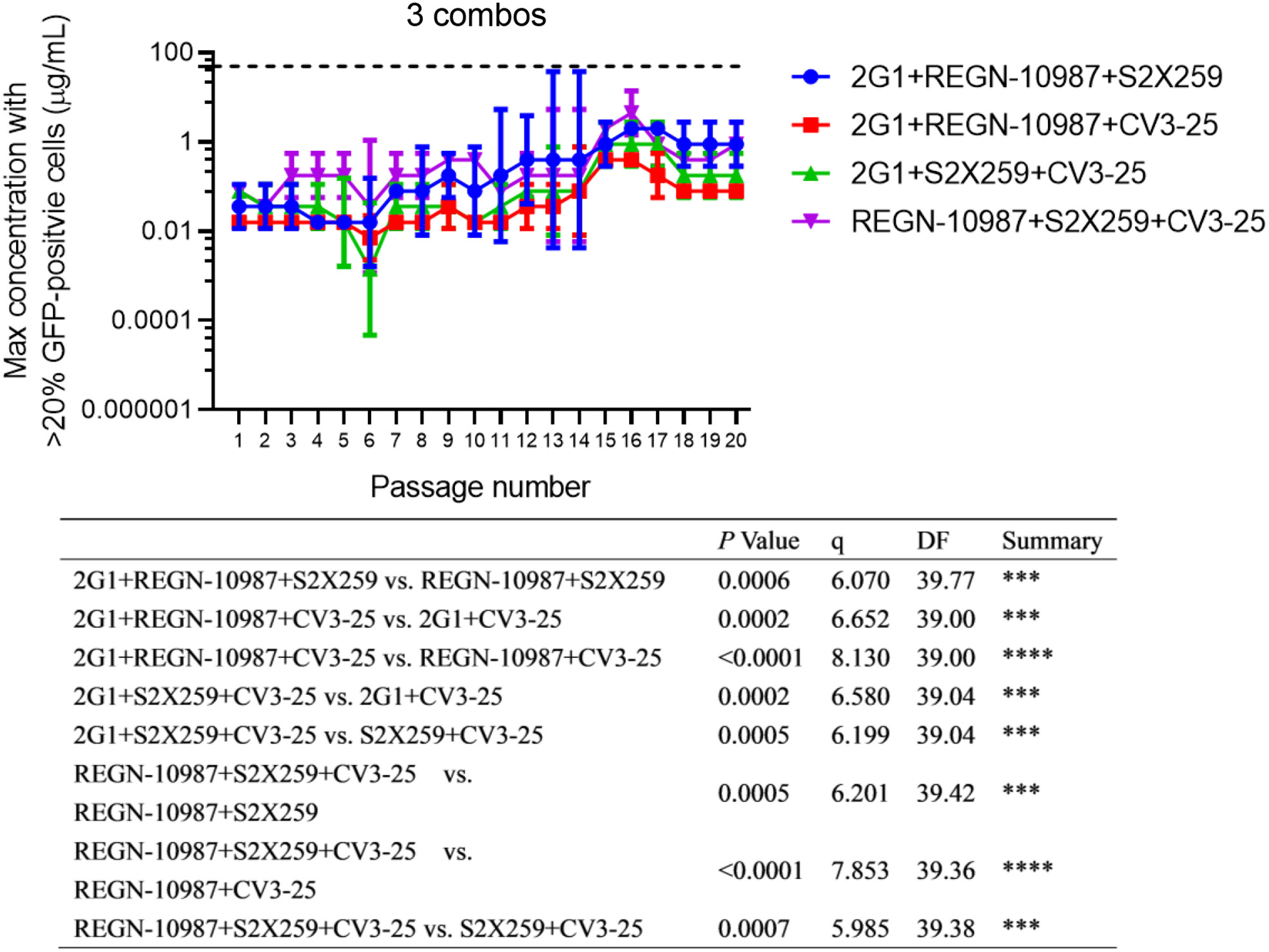
Mutational escape prevention by combination of four neutralizing antibodies that target RBD conserved regions and stem helix
2022, 37(6): 860 doi: 10.1016/j.virs.2022.11.005
Received: 23 August 2022 Accepted: 16 November 2022New variants of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) appear rapidly every few months. They have showed powerful adaptive ability to circumvent the immune system. To further understand SARS-CoV-2's adaptability so as to seek for strategies to mitigate the emergence of new variants, herein we investigated the viral adaptation in the presence of broadly neutralizing antibodies and their combinations. First, we selected four broadly neutralizing antibodies, including pan-sarbecovirus and pan-betacoronavirus neutralizing antibodies that recognize distinct conserved regions on receptor-binding domain (RBD) or conserved stem-helix region on S2 subunit. Through binding competition analysis, we demonstrated that they were capable of simultaneously binding. Thereafter, a replication-competent vesicular stomatitis virus pseudotyped with SARS-CoV-2 spike protein was employed to study the viral adaptation. Twenty consecutive passages of the virus under the selective pressure of individual antibodies or their combinations were performed. It was found that it was not hard for the virus to adapt to broadly neutralizing antibodies, even for pan-sarbecovirus and pan-betacoronavirus antibodies. The virus was more and more difficult to escape the combinations of two/three/four antibodies. In addition, mutations in the viral population revealed by high-throughput sequencing showed that under the selective pressure of three/four combinational antibodies, viral mutations were not prone to present in the highly conserved region across betacoronaviruses (stem-helix region), while this was not true under the selective pressure of single/two antibodies. Importantly, combining neutralizing antibodies targeting RBD conserved regions and stem helix synergistically prevented the emergence of escape mutations. These studies will guide future vaccine and therapeutic development efforts and provide a rationale for the design of RBD-stem helix tandem vaccine, which may help to impede the generation of novel variants. -
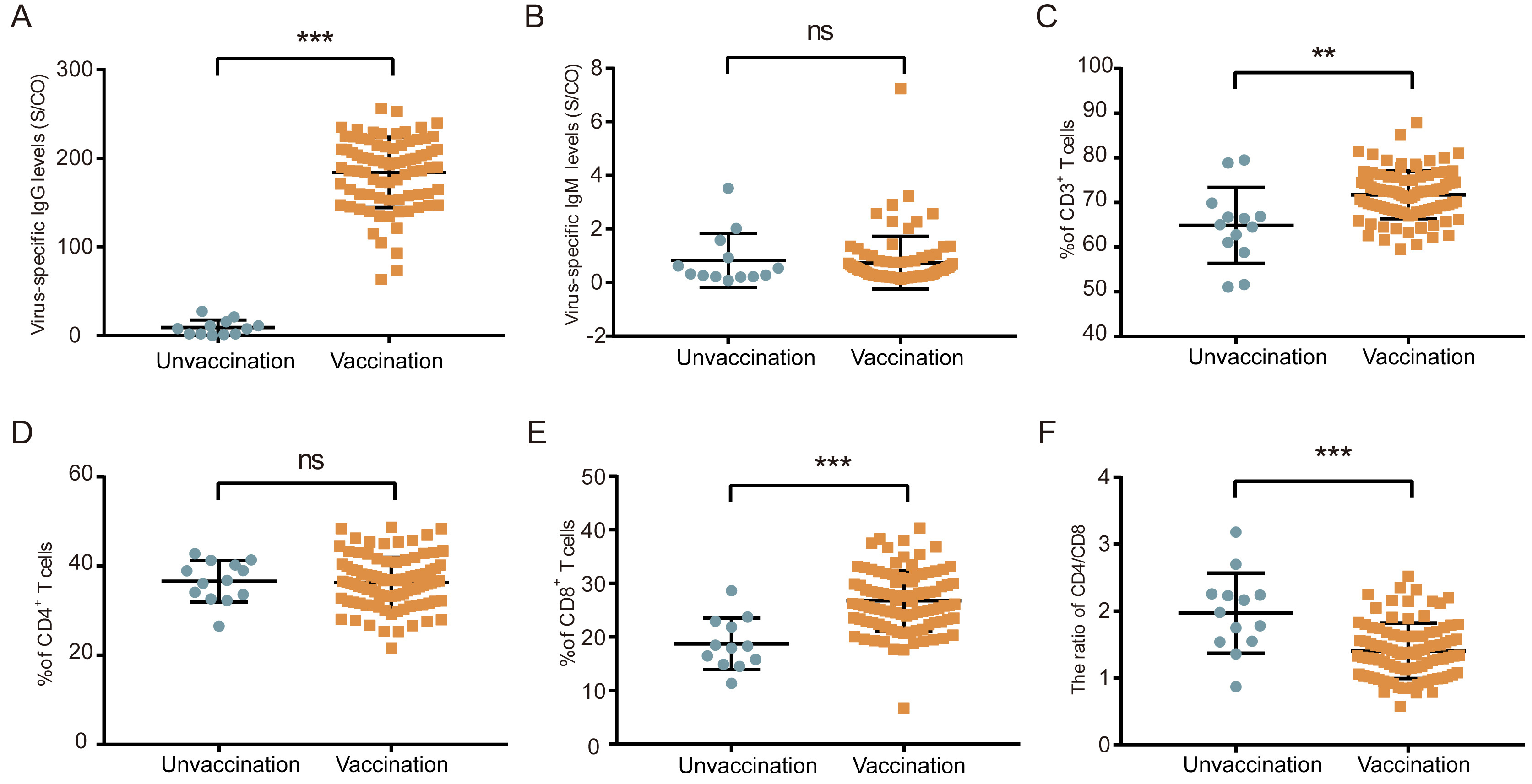
Clinical and immunological features of convalescent pediatric patients infected with the SARS-CoV-2 Omicron variant in Tianjin, China
2022, 37(6): 850 doi: 10.1016/j.virs.2022.10.009
Received: 24 April 2022 Accepted: 27 October 2022COVID-19 has spread surprisingly fast worldwide, and new variants continue to emerge. Recently, the World Health Organization acknowledged a new mutant strain “Omicron”, with children were accounting for a growing share of COVID-19 cases compared with other mutant strains. However, the clinical and immunological characteristics of convalescent pediatric patients after Omicron infection were lacking. In this study, we comparatively analyzed the clinical data from pediatric patients with adult patients or healthy children and the effects of SARSCoV-2 vaccine on the clinical and immune characteristics in convalescent pediatric patients. Our results indicated that convalescent pediatric patients had unique clinical and immune characteristics different from those of adult patients or healthy children, and SARS-CoV-2 vaccination significantly affected on the clinical and immune characteristics and the prevention of nucleic acid re-detectable positive (RP) in convalescent patients. Our study further deepens the understanding of the impact of Omicron on the long-term health of pediatric patients and provides a valuable reference for the prevention and treatment of children infected with Omicron. -
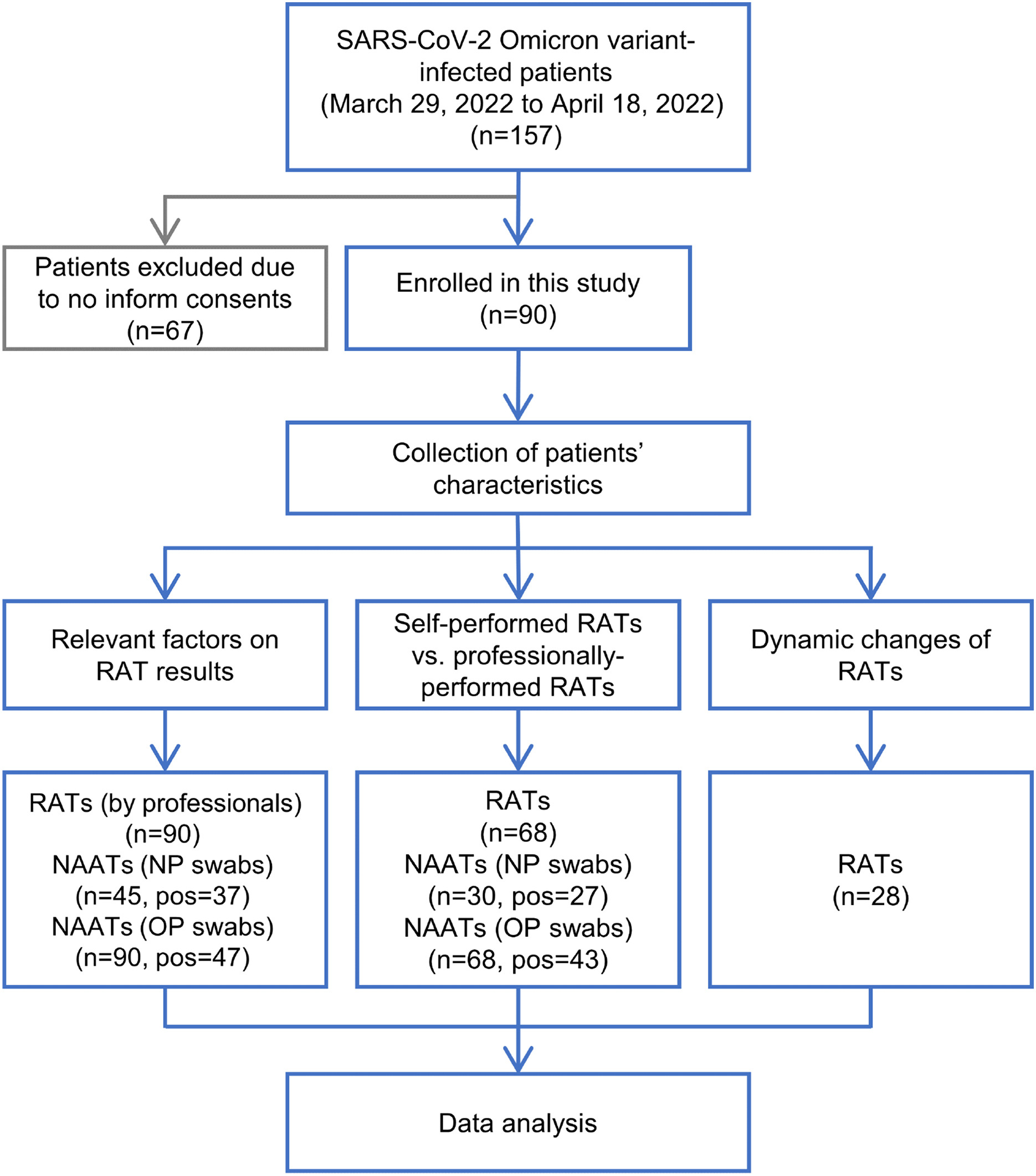
Clinical practice of rapid antigen tests for SARS-CoV-2 Omicron variant: A single-center study in China
2022, 37(6): 842 doi: 10.1016/j.virs.2022.08.008
Received: 25 May 2022 Accepted: 23 August 2022Responding to the fast-spreading SARS-CoV-2 Omicron variant, to improve screening efficiency, rapid antigen tests (RATs) were first added as a supplementary detection method in China in mid-March, 2022. What and how big a role RATs should play need to be supported by clinical data. Here, RAT performance and relevant factors in comparison with nucleic acid amplification tests (NAATs) were assessed in Omicron-infected inpatients. From the NAAT results, nasopharyngeal swabs (NPs) performed better than oropharyngeal swabs (OPs). RATs tested on NAAT positive NPs performed better than those with OP-positive samples. The RAT positivity rate was strongly associated with high levels of N and OFR1ab genes, especially in NPs where patients also had significantly longer hospital stays and shorter days from symptom onset to RAT testing. Self-performed RATs had a detection accuracy that was comparable to professionally performed RATs when the subjects were well guided. The antigen negative rate of the studied patients was 100% at discharge. These findings suggest that, in addition to a supplementary detection role, RATs can be an important strategy for evaluating the disease progression of Omicron-infected inpatients. This study provides important clinical data to support better rules regarding RATs under China's COVID-19 prevention and control policy. -
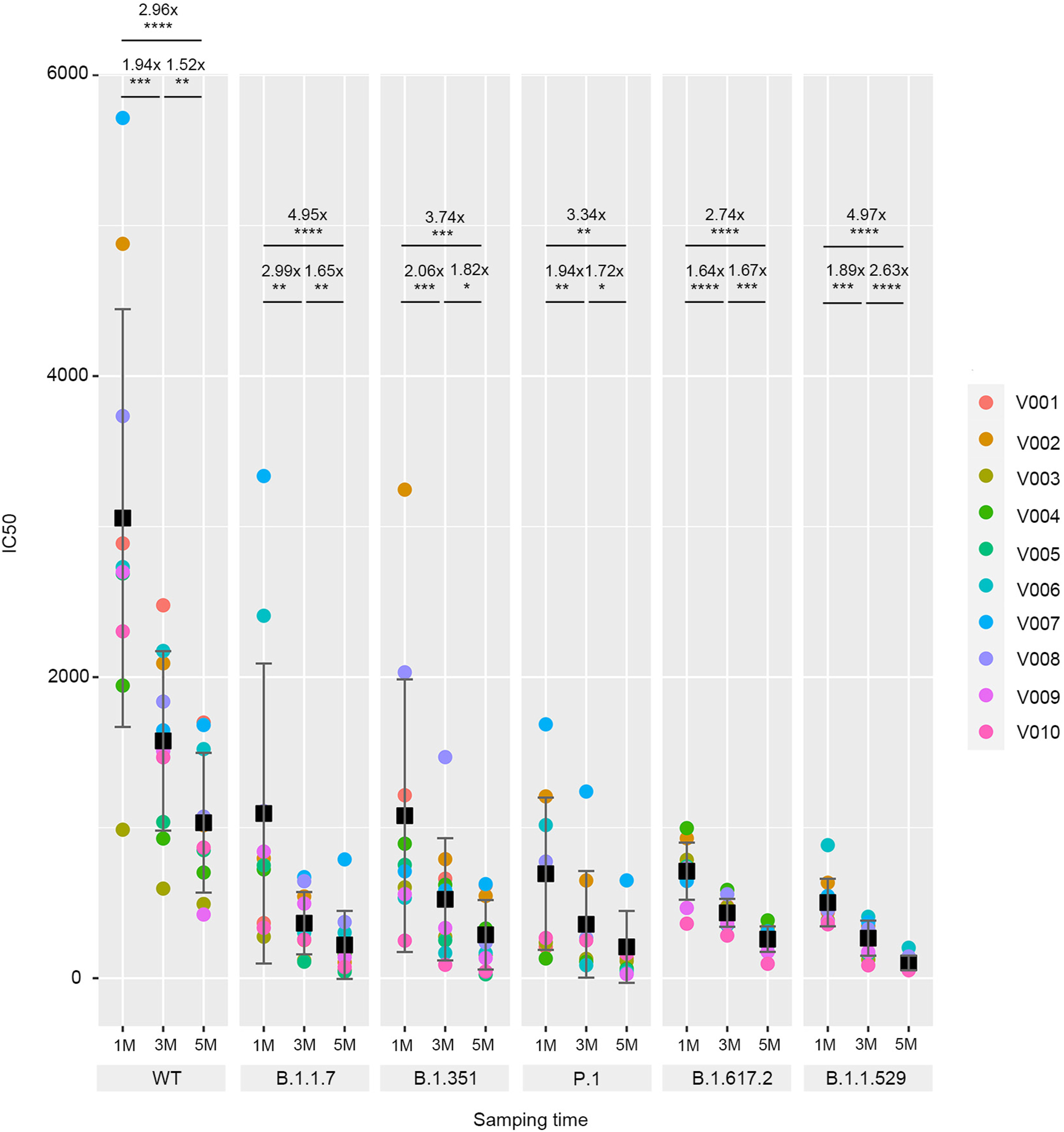
Neutralization of five SARS-CoV-2 variants of concern by convalescent and BBIBP-CorV vaccinee serum
2022, 37(6): 831 doi: 10.1016/j.virs.2022.10.006
Received: 25 July 2022 Accepted: 21 October 2022The prevalence of SARS-CoV-2 variants of concern (VOCs) is still escalating throughout the world. However, the level of neutralization of the inactivated viral vaccine recipients’ sera and convalescent sera against all VOCs, including B.1.1.7 (Alpha), B.1.351 (Beta), P.1 (Gamma), B.1.617.2 (Delta), and B.1.1.529 (Omicron) remains to be lack of comparative analysis. Therefore, we constructed pseudoviruses of five VOCs using a lentiviral-based system and analyzed their viral infectivity and neutralization resistance to convalescent and BBIBP-CorV vaccinee serum at different times. Our results show that, compared with the wild-type strain (WT), five VOC pseudoviruses showed higher infection, of which B.1.617.2 and B.1.1.529 variant pseudoviruses exhibited higher infection rates than wild-type or other VOC strains, respectively. Sera from 10 vaccinated individuals at the 1, 3 and 5-month post second dose or from 10 convalescent at 14 and 200 days after discharge retained neutralizing activity against all strains but exhibited decreased neutralization activity significantly against the five VOC variant pseudoviruses over time compared to WT. Notably, 100% (30/30) of the vaccinee serum samples showed more than a 2.5-fold reduction in neutralizing activity against B.1.1.529, and 90% (18/20) of the convalescent serum samples showed more than 2.5-fold reduction in neutralization against B.1.1.529. These findings demonstrate the reduced protection against the VOCs in vaccinated and convalescent individuals over time, indicating that it is necessary to have a booster shot and develop new vaccines capable of eliciting broad neutralization antibodies. -
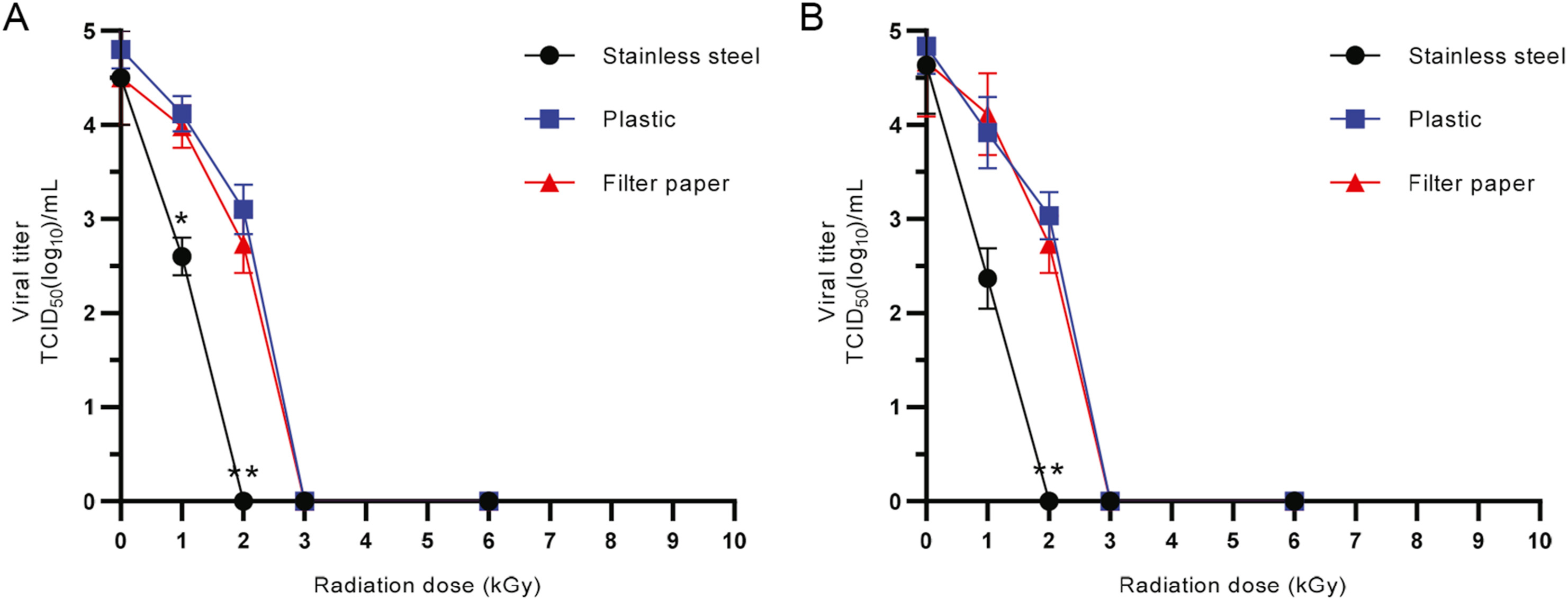
Quantitative determination of the electron beam radiation dose for SARS-CoV-2 inactivation to decontaminate frozen food packaging
2022, 37(6): 823 doi: 10.1016/j.virs.2022.10.007
Received: 01 July 2022 Accepted: 21 October 2022The spread of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) from cold-chain foods to frontline workers poses a serious public health threat during the current global pandemic. There is an urgent need to design concise approaches for effective virus inactivation under different physicochemical conditions to reduce the risk of contagion through viral contaminated surfaces of cold-chain foods. By employing a time course of electron beam exposure to a high titer of SARS-CoV-2 at cold-chain temperatures, a radiation dose of 2 kGy was demonstrated to reduce the viral titer from 104.5 to 0 median tissue culture infectious dose (TCID50)/mL. Next, using human coronavirus OC43 (HCoV-OC43) as a suitable SARS-CoV-2 surrogate, 3 kGy of high-energy electron radiation was defined as the inactivation dose for a titer reduction of more than 4 log units on tested packaging materials. Furthermore, quantitative reverse transcription PCR (RT-qPCR) was used to test three viral genes, namely, E, N, and ORF1ab. There was a strong correlation between TCID50 and RT-qPCR for SARS-CoV-2 detection. However, RT-qPCR could not differentiate between the infectivity of the radiation-inactivated and nonirradiated control viruses. As the defined radiation dose for effective viral inactivation fell far below the upper safe dose limit for food processing, our results provide a basis for designing radiation-based approaches for the decontamination of SARS-CoV-2 in frozen food products. We further demonstrate that cell-based virus assays are essential to evaluate the SARS-CoV-2 inactivation efficiency for the decontaminating strategies. -

Translation landscape of SARS-CoV-2 noncanonical subgenomic RNAs
2022, 37(6): 813 doi: 10.1016/j.virs.2022.09.003
Received: 05 July 2022 Accepted: 01 September 2022The ongoing COVID-19 pandemic is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) with a positive-stranded RNA genome. Current proteomic studies of SARS-CoV-2 mainly focus on the proteins encoded by its genomic RNA (gRNA) or canonical subgenomic RNAs (sgRNAs). Here, we systematically investigated the translation landscape of SARS-CoV-2, especially its noncanonical sgRNAs. We first constructed a strict pipeline, named vipep, for identifying reliable peptides derived from RNA viruses using RNA-seq and mass spectrometry data. We applied vipep to analyze 24 sets of mass spectrometry data related to SARS-CoV-2 infection. In addition to known canonical proteins, we identified many noncanonical sgRNA-derived peptides, which stably increase after viral infection. Furthermore, we explored the potential functions of those proteins encoded by noncanonical sgRNAs and found that they can bind to viral RNAs and may have immunogenic activity. The generalized vipep pipeline is applicable to any RNA viruses and these results have expanded the SARSCoV-2 translation map, providing new insights for understanding the functions of SARS-CoV-2 sgRNAs. -
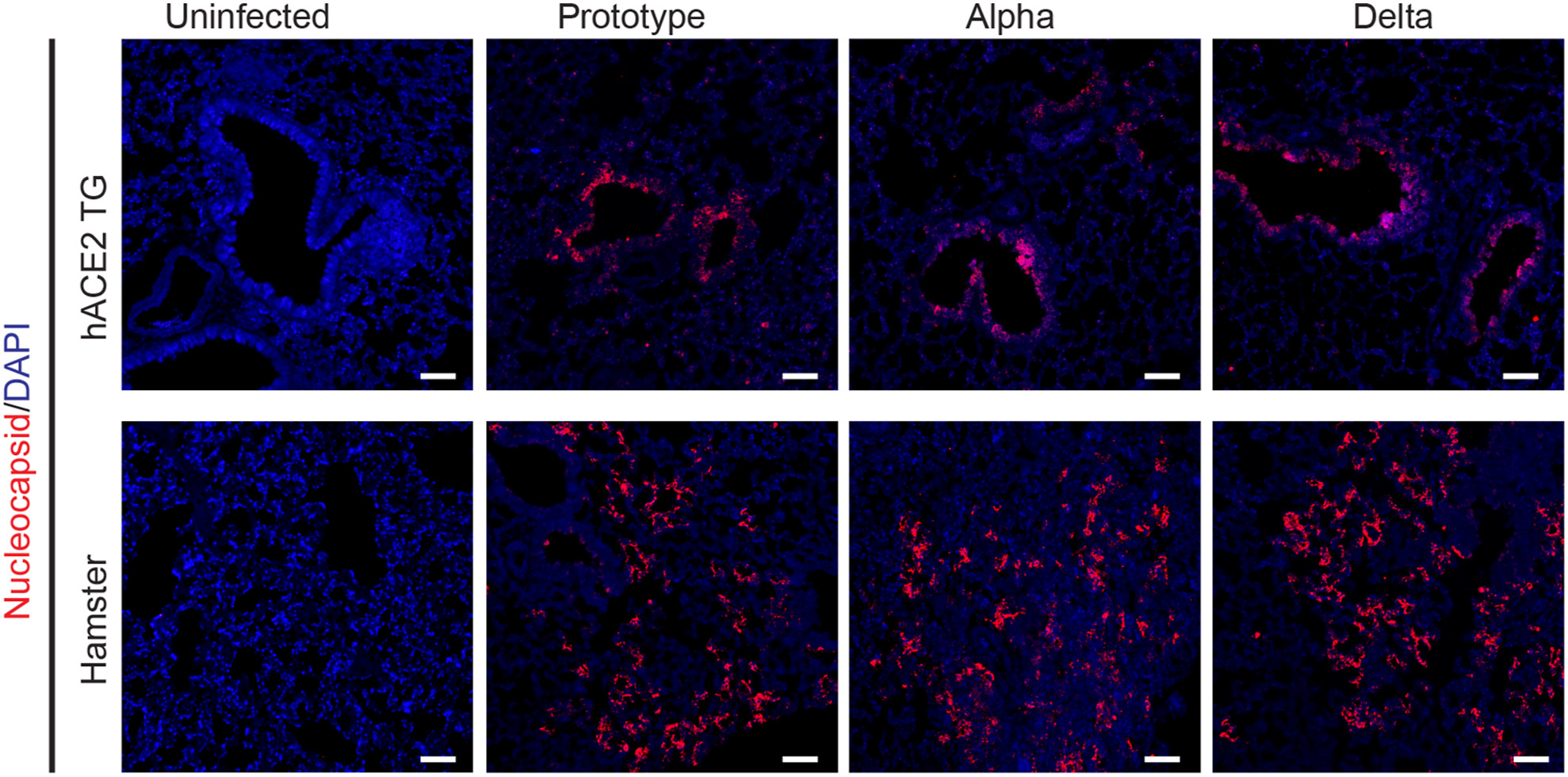
Characteristics of replication and pathogenicity of SARS-CoV-2 Alpha and Delta isolates
2022, 37(6): 804 doi: 10.1016/j.virs.2022.09.007
Received: 13 June 2022 Accepted: 20 September 2022The continuously arising of SARS-CoV-2 variants has been posting a great threat to public health safety globally, from B.1.17 (Alpha), B.1.351 (Beta), P.1 (Gamma), B.1.617.2 (Delta) to B.1.1.529 (Omicron). The emerging or reemerging of the SARS-CoV-2 variants of concern is calling for the constant monitoring of their epidemics, pathogenicity and immune escape. In this study, we aimed to characterize replication and pathogenicity of the Alpha and Delta variant strains isolated from patients infected in Laos. The amino acid mutations within the spike fragment of the isolates were determined via sequencing. The more efficient replication of the Alpha and Delta isolates was documented than the prototyped SARS-CoV-2 in Calu-3 and Caco-2 cells, while such features were not observed in Huh-7, Vero E6 and HPA-3 cells. We utilized both animal models of human ACE2 (hACE2) transgenic mice and hamsters to evaluate the pathogenesis of the isolates. The Alpha and Delta can replicate well in multiple organs and cause moderate to severe lung pathology in these animals. In conclusion, the spike protein of the isolated Alpha and Delta variant strains was characterized, and the replication and pathogenicity of the strains in the cells and animal models were also evaluated. -
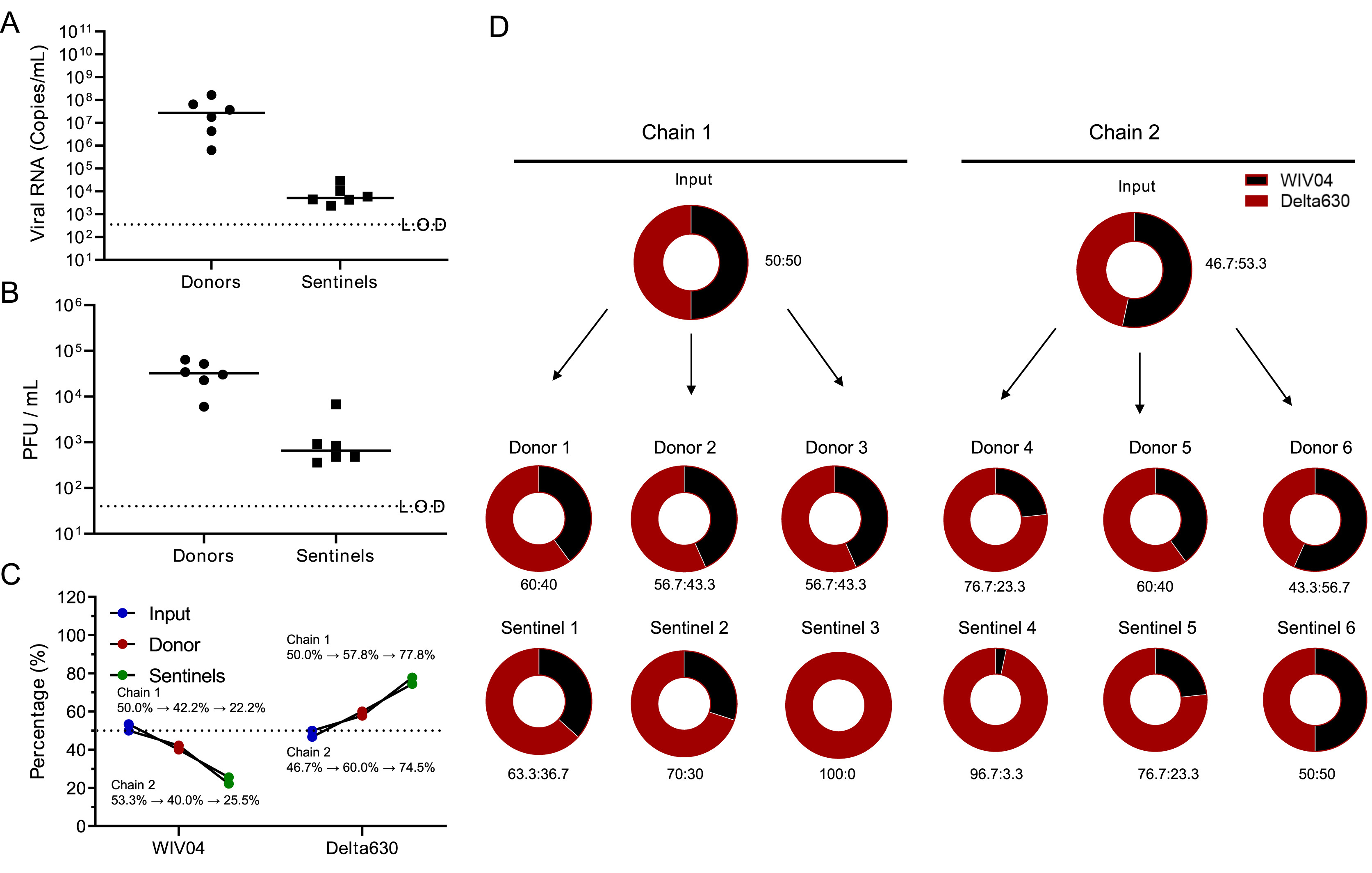
Increased pathogenicity and aerosol transmission for one SARS-CoV-2 B.1.617.2 Delta variant over the wild-type strain in hamsters
2022, 37(6): 796 doi: 10.1016/j.virs.2022.09.008
Received: 17 February 2022 Accepted: 26 September 2022During the two-year pandemic of coronavirus disease 2019 (COVID-19), its causative agent, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has been evolving. SARS-CoV-2 Delta, a variant of concern, has become the dominant circulating strain worldwide within just a few months. Here, we performed a comprehensive analysis of a new B.1.617.2 Delta strain (Delta630) compared with the early WIV04 strain (WIV04) in vitro and in vivo, in terms of replication, infectivity, pathogenicity, and transmission in hamsters. When inoculated intranasally, Delta630 led to more pronounced weight loss and more severe disease in hamsters. Moreover, 40% mortality occurred about one week after infection with 104 PFU of Delta630, whereas no deaths occurred even after infection with 105 PFU of WIV04 or other strains belonging to the Delta variant. Moreover, Delta630 outgrew over WIV04 in the competitive aerosol transmission experiment. Taken together, the Delta630 strain showed increased replication ability, pathogenicity, and transmissibility over WIV04 in hamsters. To our knowledge, this is the first SARS-CoV-2 strain that causes death in a hamster model, which could be an asset for the efficacy evaluation of vaccines and antivirals against infections of SARS-CoV-2 Delta strains. The underlying molecular mechanisms of increased virulence and transmission await further analysis. -
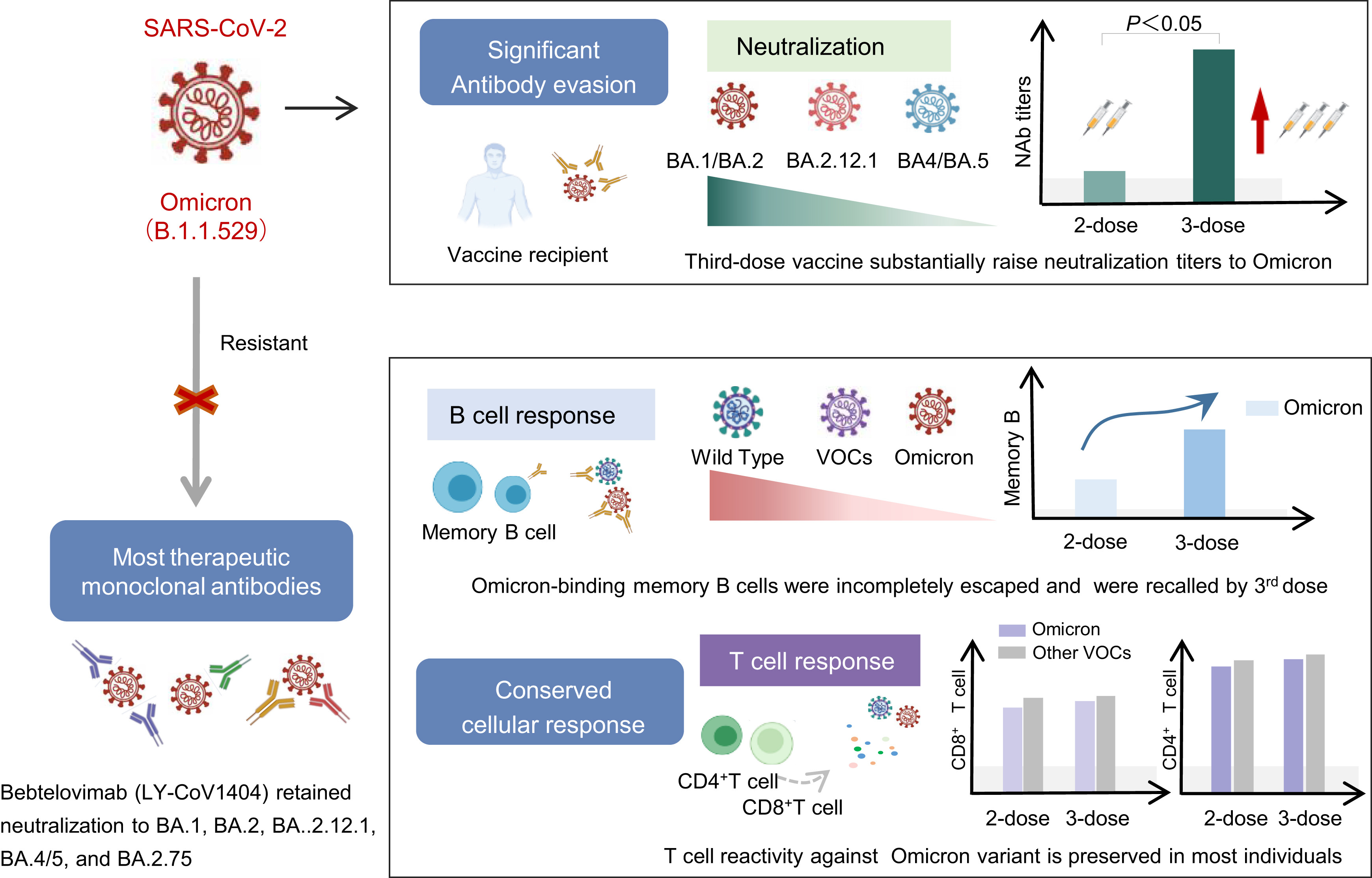
The humoral and cellular immune evasion of SARS-CoV-2 Omicron and sub-lineages
2022, 37(6): 786 doi: 10.1016/j.virs.2022.11.007
Received: 01 August 2022 Accepted: 18 November 2022The recently discovered SARS-CoV-2 variant Omicron (B.1.1.529) has rapidly become a global public health issue. The substantial mutations in the spike protein in this new variant have raised concerns about its ability to escape from pre-existing immunity established by natural infection or vaccination. In this review, we give a summary of current knowledge concerning the antibody evasion properties of Omicron and its subvariants (BA.2, BA.2.12.1, BA.4/5, and BA.2.75) from therapeutic monoclonal antibodies and the sera of SARS-CoV-2 vaccine recipients or convalescent patients. We also summarize whether vaccine-induced cellular immunity (memory B cell and T cell response) can recognize Omicron specifically. In brief, the Omicron variants demonstrated remarkable antibody evasion, with even more striking antibody escape seen in the Omicron BA.4 and BA.5 sub-lineages. Luckily, the third booster vaccine dose significantly increased the neutralizing antibodies titers, and the vaccine-induced cellular response remains conserved and provides second-line defense against the Omicron.

- First
- Prev
- 1
- 2
- Next
- Last
- Total:2
- To
- Go