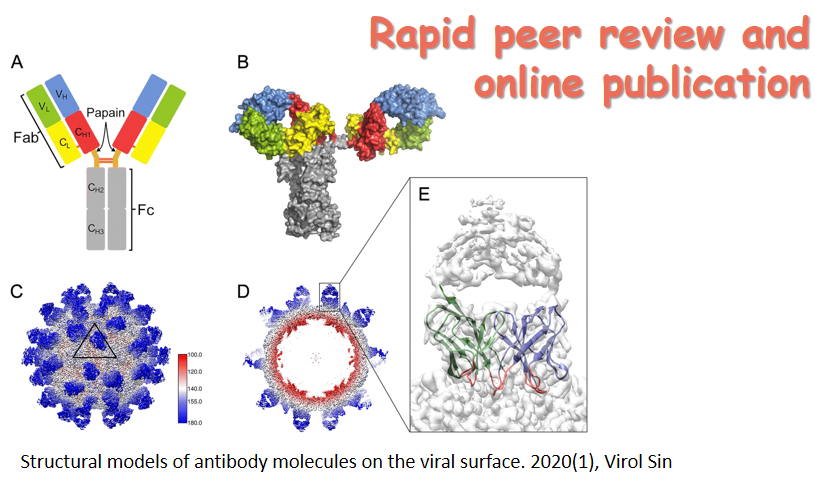
Bats have been recognized as an important natural reservoir for zoonotic viruses affecting both humans and livestock. They are likely to shed deadly emerging pathogens through feces and urine, materials that can be highly infectious to humans. In this issue, Waruhiu et al. performed the first country-wide surveillance of bat-borne viruses in Kenya spanning from 2012–2015 covering sites perceived to have medium to high level bat-human interaction. Nine virus families were screened, and 8 were detected with varying distributions. The study provides new and extended knowledge with regards to the virus diversity in Kenyan bat species, and hence raises the public health concern on the importance of continuous surveillance (See page 101-114 for details). The cover is a photo captured during the investigation in a bat cave in Kenya. (Image courtesy of Prof. Zhengli Shi).
Cecilia Waruhiu, Sheila Ommeh, Vincent Obanda, Bernard Agwanda, Francis Gakuya, Xing-Yi Ge, Xing-Lou Yang, Li-Jun Wu, Ali Zohaib, Ben Hu and Zheng-Li Shi. Molecular detection of viruses in Kenyan bats and discovery of novel astroviruses, caliciviruses and rotaviruses[J]. Virologica Sinica, 2017, 32(2): 101-114. doi: 10.1007/s12250-016-3930-2.
This is the first country-wide surveillance of bat-borne viruses in Kenya spanning from 2012-2015 covering sites perceived to have medium to high level bat-human interaction.The objective of this surveillance study was to apply a non-invasive approach using fresh feces to detect viruses circulating within the diverse species of Kenyan bats.We screened for both DNA and RNA viruses; specifically,astroviruses (AstVs),adenoviruses (ADVs),caliciviruses (CalVs),coronaviruses (CoVs),flaviviruses,filoviruses,paramyxoviruses (PMVs),polyomaviruses (PYVs) and rotaviruses. We used family-specific primers,amplicon sequencing and further characterization by phylogenetic analysis.Except for filoviruses,eight virus families were detected with varying distributions and positive rates across the five regions (former provinces) studied.AstVs (12.83%),CoVs (3.97%),PMV (2.4%),ADV (2.26%),PYV (1.65%),CalVs (0.29%),rotavirus (0.19%) and flavivirus (0.19%).Novel CalVs were detected in Rousettus aegyptiacus and Mops condylurus while novel Rotavirus-A-related viruses were detected in Taphozous bats and R.aegyptiacus.The two Rotavirus A (RVA) strains detected were highly related to human strains with VP6 genotypes I2 and I16.Genotype I16 has previously been assigned to human RVA-strain B10 from Kenya only,which raises public health concern,particularly considering increased human-bat interaction. Additionally,229E-like bat CoVs were detected in samples originating from Hipposideros bats roosting in sites with high human activity.Our findings confirm the presence of diverse viruses in Kenyan bats while providing extended knowledge on bat virus distribution.The detection of viruses highly related to human strains and hence of public health concern,underscores the importance of continuous surveillance.
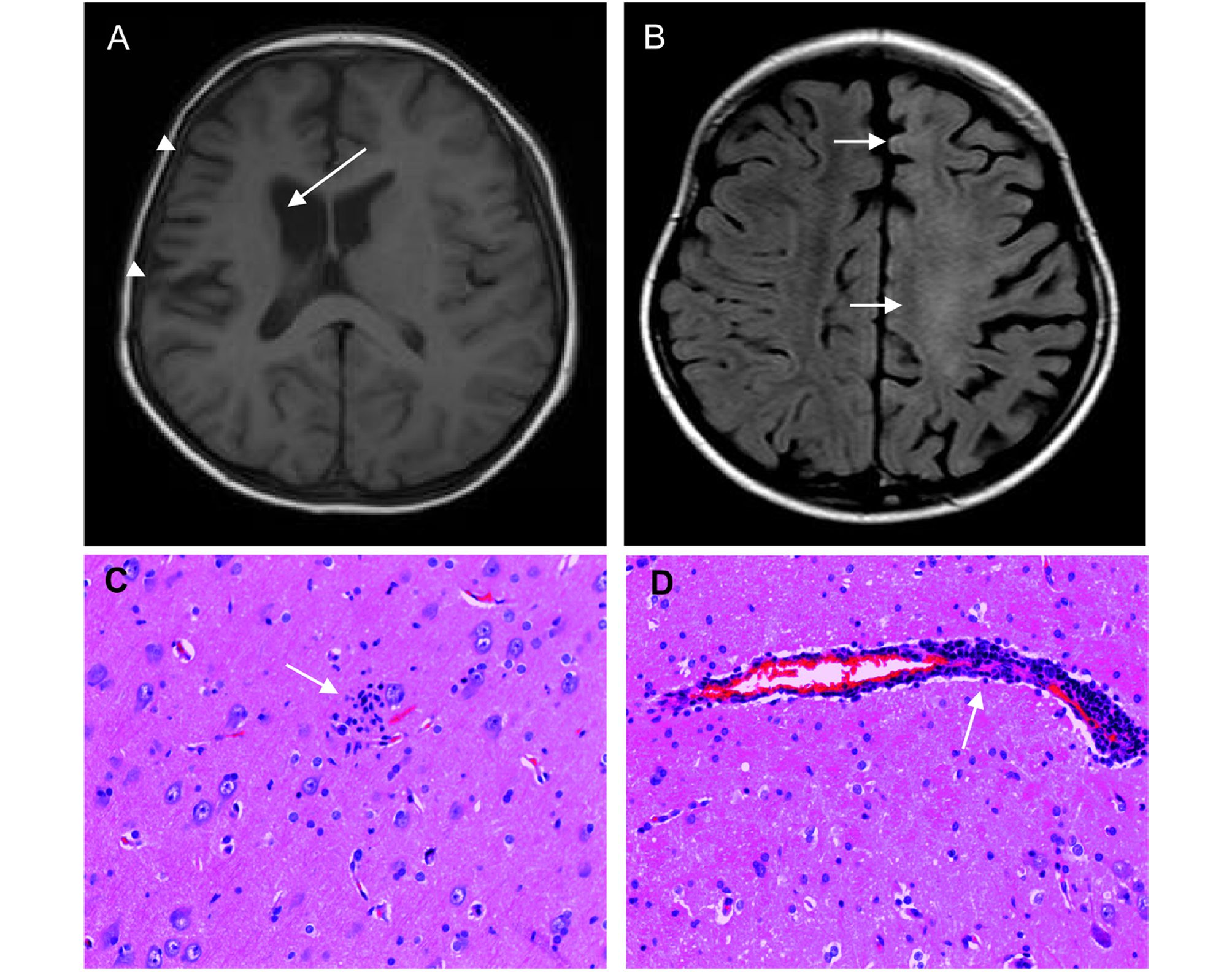
Yao Zhang, Yisong Wang, Sichang Chen, Shuai Chen, Yuguang Guan, Yuguang Guan, Changqing Liu, Tianfu Li, Guoming Luan and Jing An. Expression of human cytomegalovirus components in the brain tissues of patients with Rasmussen's encephalitis[J]. Virologica Sinica, 2017, 32(2): 115-121. doi: 10.1007/s12250-016-3917-z.
Rasmussen's encephalitis (RE) is a rare and severe progressive epileptic syndrome with unknown etiology.Infection by viruses,including human cytomegalovirus (HCMV),has been speculated to be a potential trigger for RE.However,no viral antigens have been detected in the brains of patients with RE;thus,a possible clinical linkage between viral infections and RE has not been firmly established.In this study,we evaluated the expression of HCMV pp65 antigen in brain sections from 26 patients with RE and 20 non-RE patients by immunohistochemistry and in situ hybridization,and assessed the associations between HCMV infection and clinical parameters. Elevated expression of HCMV pp65 protein and DNA was observed in 88.5%(23/26) and 69.2% (18/26) of RE cases,respectively.In the non-RE group,HCMV pp65 antigen was detected only in two cases (10%),both of which were negative for DNA staining.Additionally,the intensity of HCMV pp65 staining was correlated with a shorter duration of the prodromal stage,younger age of seizure onset,and more severe unilateral cortical atrophy.Elevated expression of HCMV pp65 was observed in RE brain tissue and was correlated with the clinical features of RE disease.In summary,our results suggested that HCMV infection may be involved in the occurrence and progression of RE disease.Thus,further studies are needed to determine whether early treatment with anti-HCMV antibodies could modulate the course of RE.
Mao Huawei, Yinping Liu, Sin Fun Sia, JS Malik Peiris, Yu-Lung Lau and Tu Wenwei. Avian influenza virus directly infects human natural killer cells and inhibits cell activity[J]. Virologica Sinica, 2017, 32(2): 122-129. doi: 10.1007/s12250-016-3918-y.
Natural killer (NK) cell is a key component of innate immunity and plays an important role in host defense against virus infection by directly destroying infected cells.Influenza is a respiratory disease transmitted in the early phase of virus infection.Evasion of host innate immunity including NK cells is critical for the virus to expand and establish a successful acute infection.Previously, we showed that human influenza H1N1 virus infects NK cells and induces cell apoptosis,as well as inhibits NK cell activity.In this study,we further demonstrated that avian influenza virus also directly targeted NK cells as an immunoevasion strategy.The avian virus infected human NK cells and induced cell apoptosis.In addition,avian influenza virion and HA protein inhibited NK cell cytotoxicity.This novel strategy has obvious advantages for avian influenza virus,allowing the virus sufficient time to expand and subsequent spread before the onset of the specific immune response.Our findings provide an important clue for the immunopathogenesis of avian influenza, and also suggest that direct targeting NK cells may be a common strategy used by both human and avian influenza viruses to evade NK cell immunity.
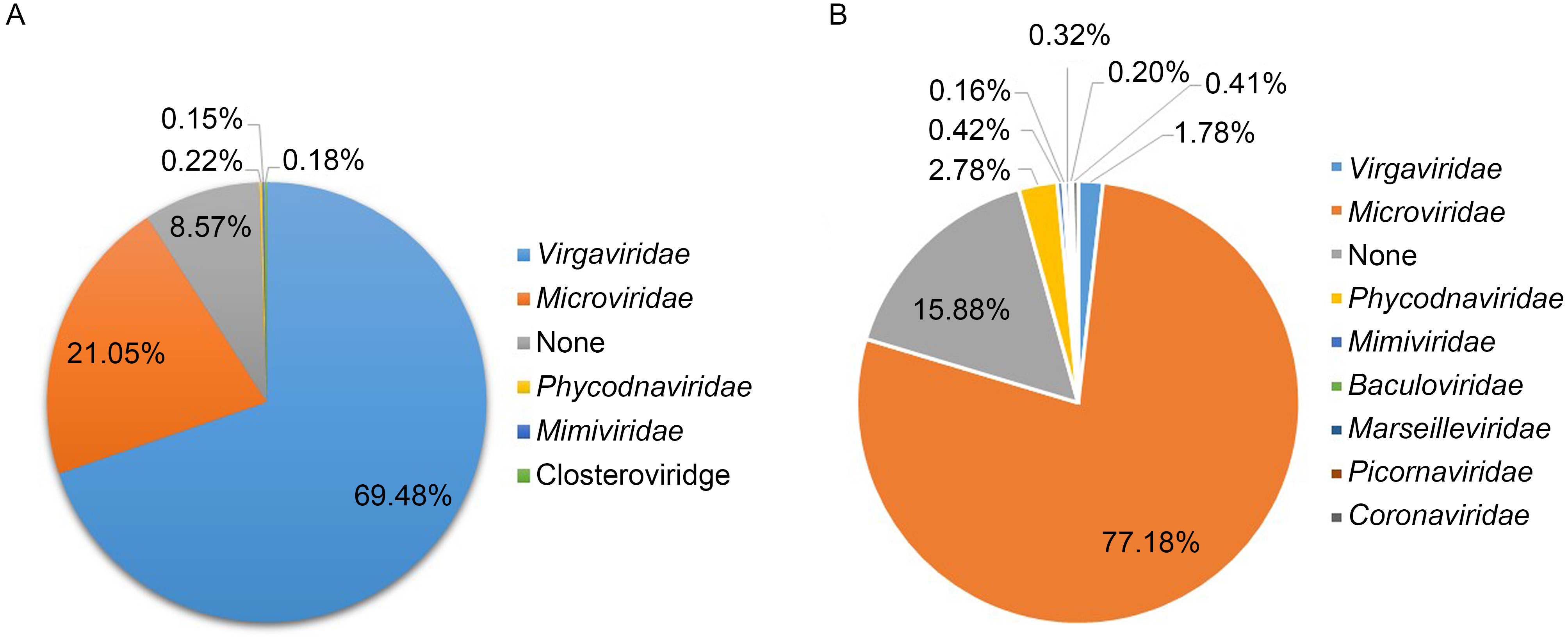
Lianghua Guo, Xiuguo Hua, Wen Zhang, Shixing Yang, Quan Shen, Haibing Hu, Jingjiao Li, Zhijian Liu, Xiaochun Wang, Hua Wang, Chenglin Zhou and Li Cui. Viral metagenomics analysis of feces from coronary heart disease patients reveals the genetic diversity of the Microviridae[J]. Virologica Sinica, 2017, 32(2): 130-138. doi: 10.1007/s12250-016-3896-0.
Recent studies have declared that members of the ssDNA virus family Microviridae play an important role in multiple environments,as they have been found taking a dominant position in the human gut.The aim of this study was to analyze the overall composition of the gut virome in coronary heart disease (CHD) patients,and try to discover the potential link between the human gut virome and CHD.Viral metagenomics methods were performed to detect the viral sequences in fecal samples collected from CHD inpatients and healthy persons as controls.We present the analysis of the virome composition in these CHD patients and controls.Our data shows that the virome composition may be linked to daily living habits and the medical therapy of CHD. Virgaviridae and Microviridae were the two dominant types of viruses found in the enteric virome of CHD patients.Fourteen divergent viruses belonging to the family Microviridae were found,twelve of which were grouped into the subfamily Gokushovirinae,while the remaining two strains might represent two new subfamilies within Microviridae,according to the phylogenetic analysis.In addition,the genomic organization of these viruses has been characterized.
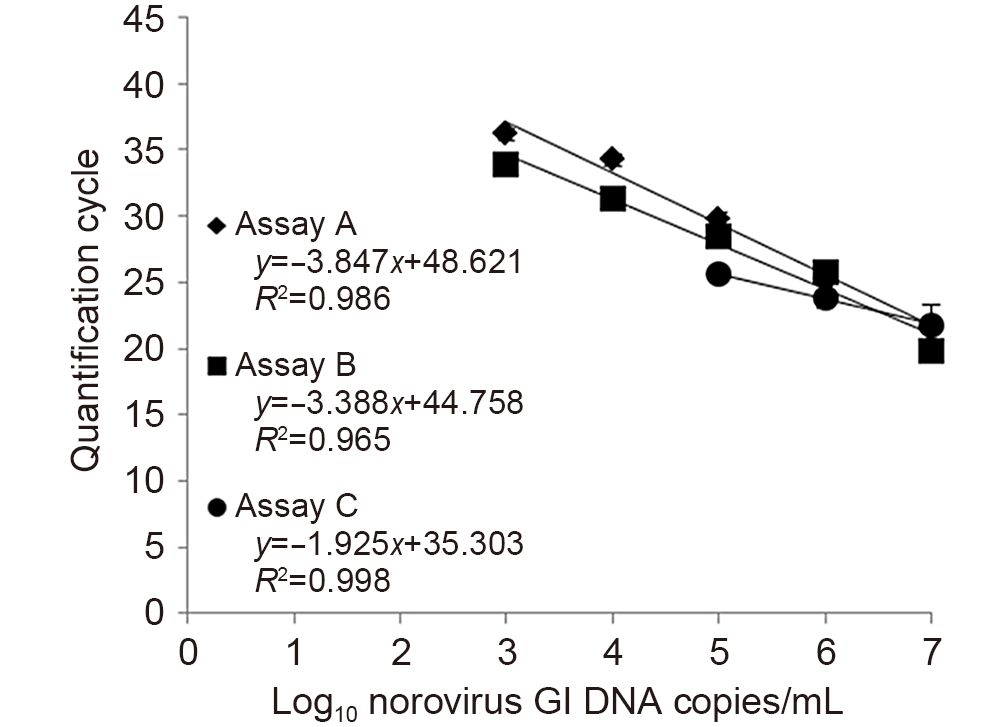
Kitwadee Rupprom, Porntip Chavalitshewinkoon-Petmitr, Pornphan Diraphat and Leera Kittigul. Evaluation of real-time RT-PCR assays for detection and quantification of norovirus genogroups Ⅰ and Ⅱ[J]. Virologica Sinica, 2017, 32(2): 139-146. doi: 10.1007/s12250-016-3863-9.
Noroviruses are the leading cause of acute gastroenteritis in humans.Real-time reverse transcription-polymerase chain reaction (real-time RT-PCR) is a promising molecular method for the detection of noroviruses.In this study,the performance of three TaqMan real-time RT-PCR assays was assessed,which were one commercially available real-time RT-PCR kit (assay A: Norovirus Real Time RT-PCR kit) and two in-house real-time RT-PCR assays (assay B:LightCycler RNA Master Hybprobe and assay C:RealTime ready RNA Virus Master).Assays A and B showed higher sensitivity than assay C for norovirus GI,while they all had the same sensitivity (103 DNA copies/mL) for GII DNA standard controls.Assay B had the highest efficiency for both genogroups. No cross-reactivity was observed among GI and GII noroviruses,rotavirus,hepatitis A virus,and poliovirus.The detection rates of these assays in GI and GII norovirus-positive fecal samples were not significantly different.However,the mean quantification cycle (Cq) value of assay B for GII was lower than assays A and C with statistical significance (P-value,0.000).All three real-time RT-PCR assays could detect a variety of noroviruses including GI.2,GII.2,GII.3,GII.4,GII.6,GII.12,GII.17, and GII.21.This study suggests assay B as a suitable assay for the detection and quantification of noroviruses GI and GII due to good analytical sensitivity and higher performance to amplify norovirus on DNA standard controls and clinical samples.
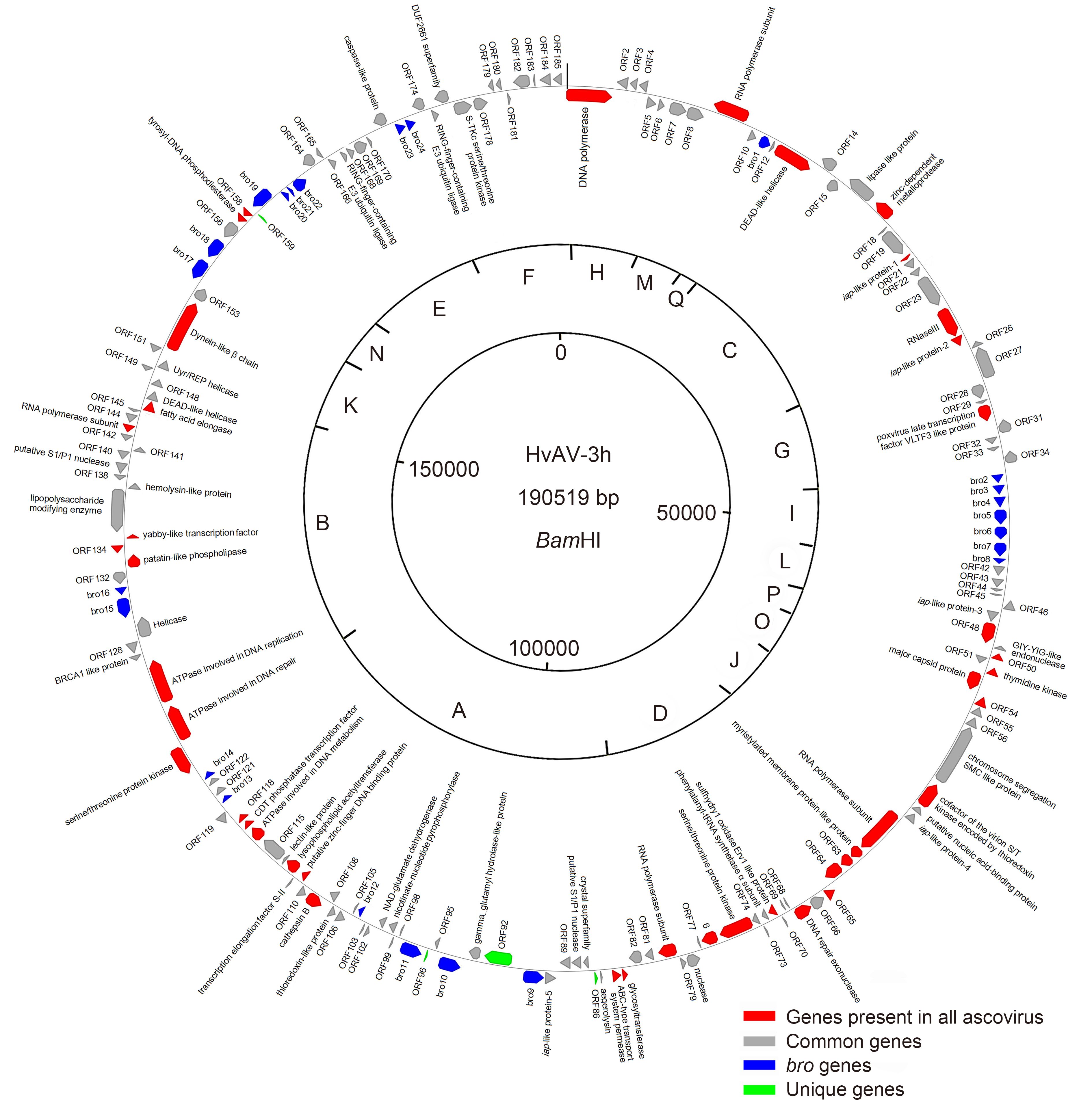
Guo-Hua Huang, Dian-Hai Hou, Manli Wang, Xiao-Wen Cheng and Zhihong Hu. Genome analysis of Heliothis virescens ascovirus 3h isolated from China[J]. Virologica Sinica, 2017, 32(2): 147-154. doi: 10.1007/s12250-016-3929-8.
No ascovirus isolated from China has been sequenced so far.Therefore,in this study,we aimed to sequence the genome of Heliothis virescens ascovirus 3h (HvAV-3h) using the 454 pyrosequencing technology.The genome was found to be 190,519-bp long with a G+C content of 45.5%.We also found that it encodes 185 hypothetical open reading frames (ORFs) along with at least 50 amino acids,including 181 ORFs found in other ascoviruses and 4 unique ORFs.Gene-parity plots and phylogenetic analysis revealed a close relationship between HvAV-3h and three other HvAV-3a strains and a distant relationship with Spodoptera frugiperda ascovirus 1a (SfAV-1a),Trichoplusia ni ascovirus 6a (TnAV-6a),and Diadromus pulchellus ascovirus 4a (DpAV-4a).Among the 185 potential genes encoded by the genome,44 core genes were found in all the sequenced ascoviruses.In addition,25 genes were found to be conserved in all ascoviruses except DpAV-4a. In the HvAV-3h genome,24 baculovirus repeat ORFs (bros) were present,and the typical homologous repeat regions (hrs) were absent.This study supplies information important for understanding the conservation and functions of ascovirus genes as well as the variety of ascoviral genomes.
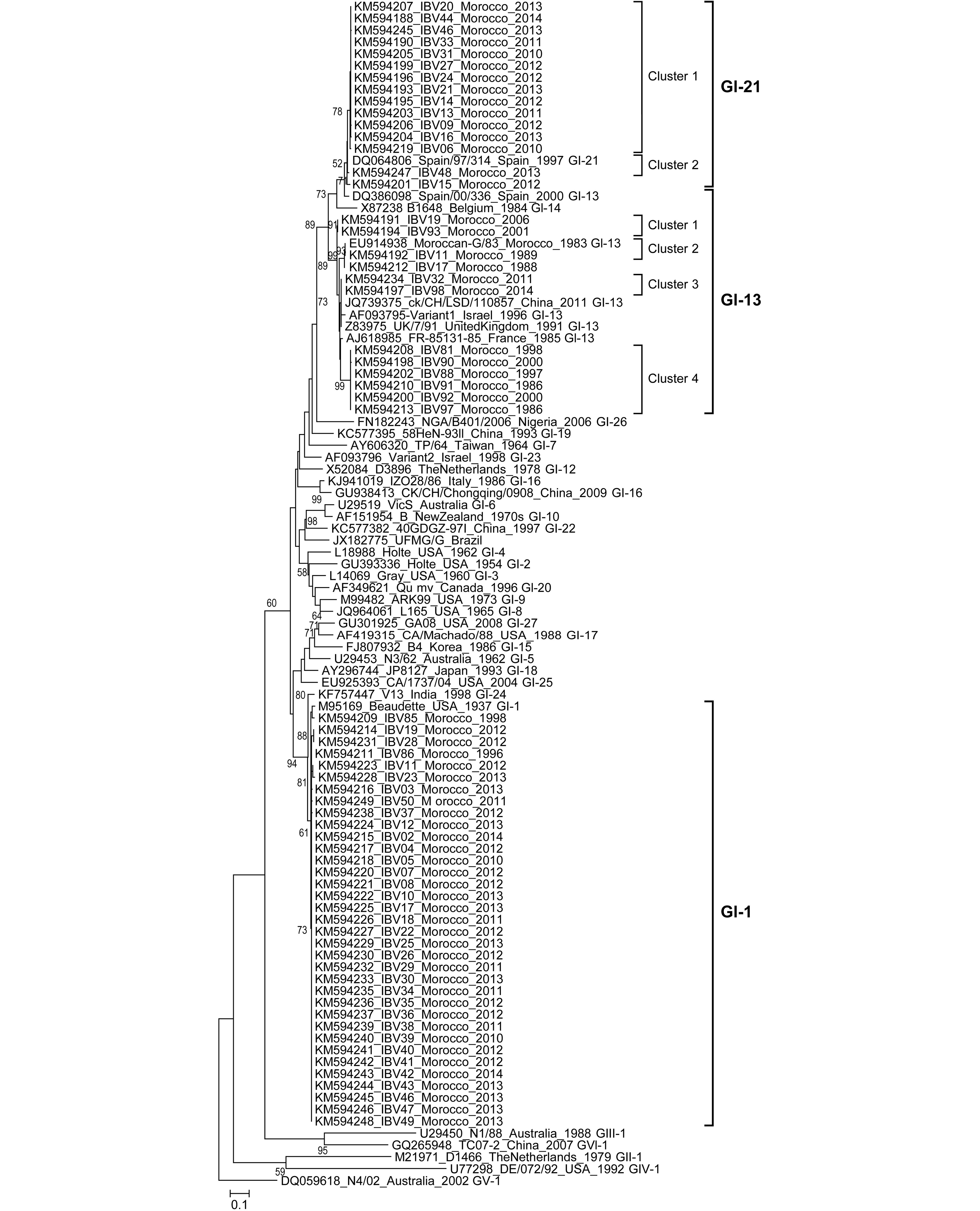
Siham Fellahi, Mehdi El Harrak, Slimane Khayi, Jean-Luc Guerin, Jens H. Kuhn, Mohammed El Houadfi, My Mustapha Ennaji and Mariette Ducatez. Phylogenetic analysis of avian infectious bronchitis virus isolates from Morocco: a retrospective study (1983 to 2014)[J]. Virologica Sinica, 2017, 32(2): 155-158. doi: 10.1007/s12250-016-3885-3.
In Morocco, molecular and genetic characterization of IBV strain diversity have been very limited. A long-term retrospective study on Moroccan isolates is required to improve understanding of IBV evolution and IB epidemiology. The objective of this report is to perform a retrospective analysis of the origin and evolution of 62 Moroccan IBV isolates obtained from unvaccinated (21%) and vaccinated (70%) poultry flocks showing clinical signs of IB between 1983 and 2014.
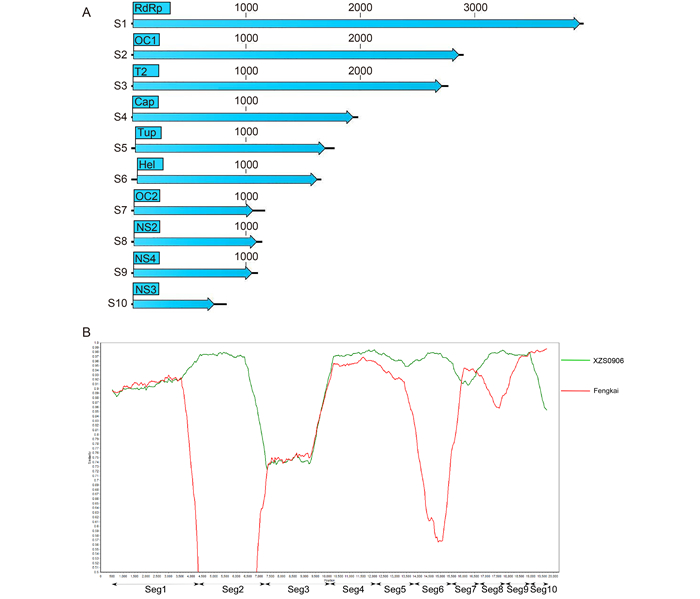
Shaozhen Xing, Xiaofang Guo, Xianglilan Zhang, Qiumin Zhao, Lingli Li, Shuqing Zuo, Xiaoping An, Guangqian Pei, Qiang Sun, Shi Cheng, Yunfei Wang, Hang Fan, Zhiqiang Mi, Yong Huang, Zhiyi Zhang, Hongning Zhou, Jiusong Zhang and Yigang Tong. A novel mosquito-borne reassortant orbivirus isolated from Xishuangbanna, China[J]. Virologica Sinica, 2017, 32(2): 159-162. doi: 10.1007/s12250-016-3886-2.
In this study, we report a novel mosquito-borne reassortant Orbivirus species, which we named Banna orbivirus (BAOV). The virus was isolated from a Culex tritaeniorhynchus pool collected in Xishuangbanna, Yunnan Province, China in 2007. Whole genome and phylogenetic analyses revealed that BAOV is a reassortant of several Tibet orbivirus strains. It has a double-stranded RNA genome of 19, 270 bp, containing ten segments (S1–S10) of various lengths. The 10 segments of BAOV had high nucleotide and amino acid sequence similarity with segments of either Fengkai orbivirus or Tibet orbivirus (TIBOV, XZ0906 strain). Phylogenetic analyses indicated that the novel BAOV is a reassortant derived from XZ0906 and Fengkai orbiviruses, and is a Tibet orbivirus. This is the first report of a novel genomic reassortment of Tibet orbivirus isolated in China; these findings contribute to our understanding of the diversity of orbiviruses.
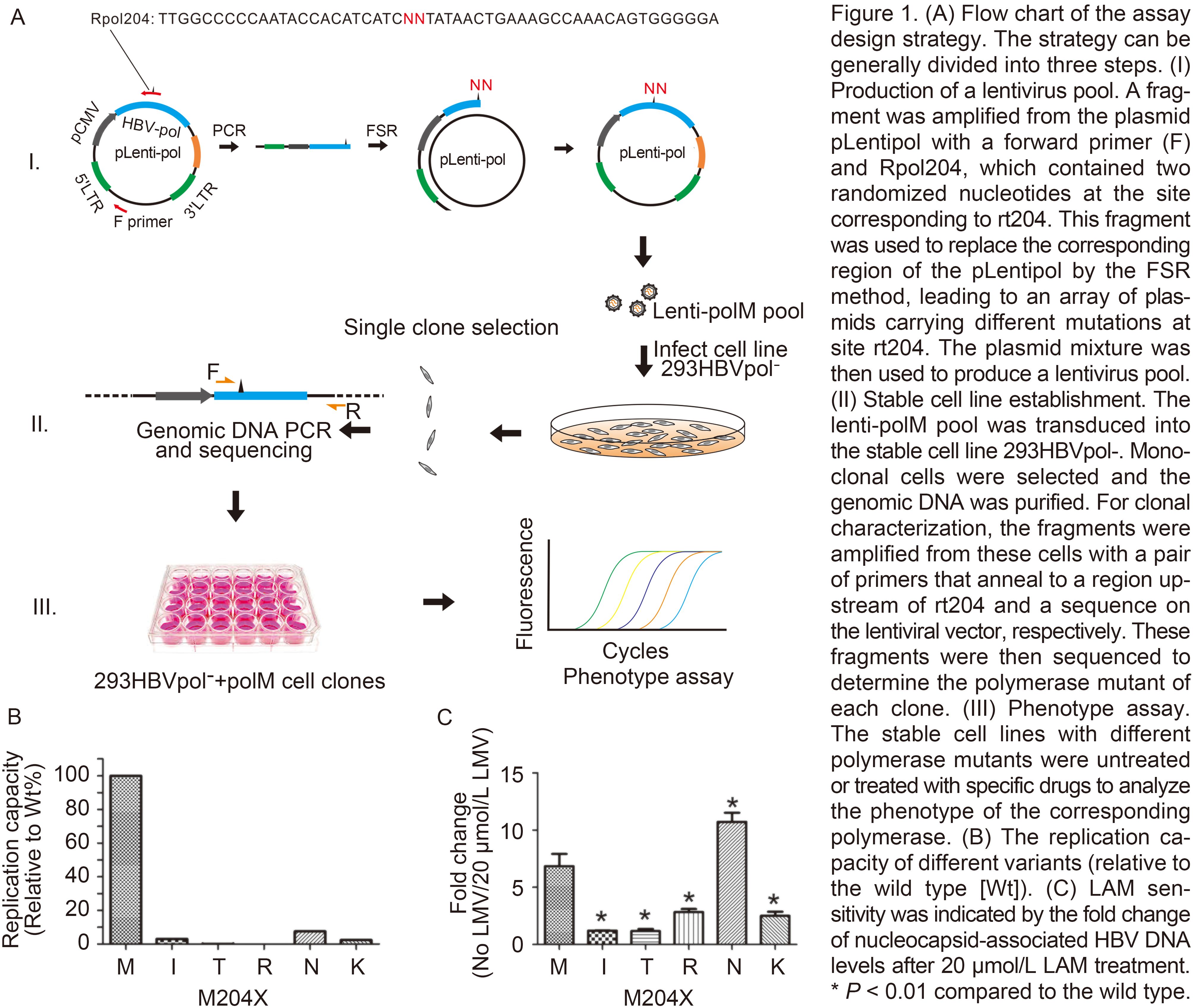
Fuxian Zhang, Shicui Yan, Hong Guo, Qingxiu Chen and Fang Qin. Characterization of viral entry and infection of quantum dotlabeled grass carp reovirus[J]. Virologica Sinica, 2017, 32(2): 163-166. doi: 10.1007/s12250-016-3903-5.
In this study, we characterized a QD-labeled nonenveloped aquareovirus. Our results showed that QDGCRVs maintained their native particle properties and showed excellent infectivity in host cells. To the best of our knowledge, this is the first study reporting the realization of in situ real-time tracking with fluorophores in live cells by utilizing a QD-labeled non-enveloped aquareovirus. Although the QD-GCRV can’t track the whole process of viral infection, this approach will provide a promising prospect for further studies of the molecular mechanisms of viral entry into host cells during aquareovirus infection.
Ya Liu, Ying-Ying Luo, Xue-Fei Cai, Quan-Xin Long, Chun-Yang Gan, Liu-Qing Yang, Haitao Guo, Ai-Long Huang, Wen-Lu Zhang and Jie-Li Hu. A novel phenotypic assay of hepatitis B virus polymerase with extensive site-specific mutagenesis[J]. Virologica Sinica, 2017, 32(2): 167-170. doi: 10.1007/s12250-016-3932-0.
In summary, we developed a novel mutagenesis phenotypic assay with the following characteristics: (1) acquisition of extensive site-specific mutations through a PCR step and FSR; (2) production of a lentivirus pool expressing different polymerase mutants via one-time lentivirus packaging; (3) generation of stable cell lines that replicate HBV DNA by integrated polymerase mutants, which can help to establish an experimental system with lower variation and higher convenience. This randomized phenotypic assay of HBV polymerase provides a useful tool for assessing the replication fitness and drug susceptibility of HBV variants and quasi-species, which may offer new insight for the NUC therapy and the prognosis of patients with chronic hepatitis B.
Dongmei Chen, Qinwei Song, Runan Zhu, Yuan Qian, Yu Sun, Jie Deng, Fang Wang, Yaxin Ding, Run Tian, Chuanhe Liu, Wenjing Zhu and Linqing Zhao. Human rhinovirus C infection is associated with asthma in children determined by xTAG respiratory viral panel FAST[J]. Virologica Sinica, 2017, 32(2): 171-174. doi: 10.1007/s12250-016-3935-x.
Cumulative evidence supports the role of early-life viral infections, especially respiratory syncytial virus (RSV) and human rhinovirus (HRV), as major antecedents of childhood asthma (Lemanske, 2002; Jackson et al.,2008). In this study, the xTAG respiratory viral panel FAST (RVP FAST) assay, a multiplex polymerase chain reaction (PCR)-based method (Arens et al., 2010; BaladaLlasat et al., 2011; Gharabaghi et al., 2011; Selvaraju, 2012), was used to investigate the association of infection with different HRV species, especially HRV-C, and the development of asthma in young children living in Beijing, China.