With decades of efforts and steady progress, Virologica Sinica has become one of high-quality academic journals in Virology with an impact factor 4.327, based on the newly released JCR®2020. Virologica Sinica switches to fully open access in 2022 for faster publishing, greater visibility, and wider connection. Once the article is accepted, it will be directly published in the form of pre-proof on the ScienceDirect domain of Elsevier, the host platform of Virologica Sinica's publishing partner KeAi Communications.
Aiming for a leading journal in Virology, we will continue to serve the scientific community actively and dedicatedly, strive to be the trusted journal that virology scientists constantly choose to publish their important work, and inspire future virology and pertaining multidiscipline researches.
A set of pathogenic human viruses, including SARS-CoV-2, HIV-1, influenza viruses, flaviviruses, Ebola viruses, are artistically presenting on the cover.
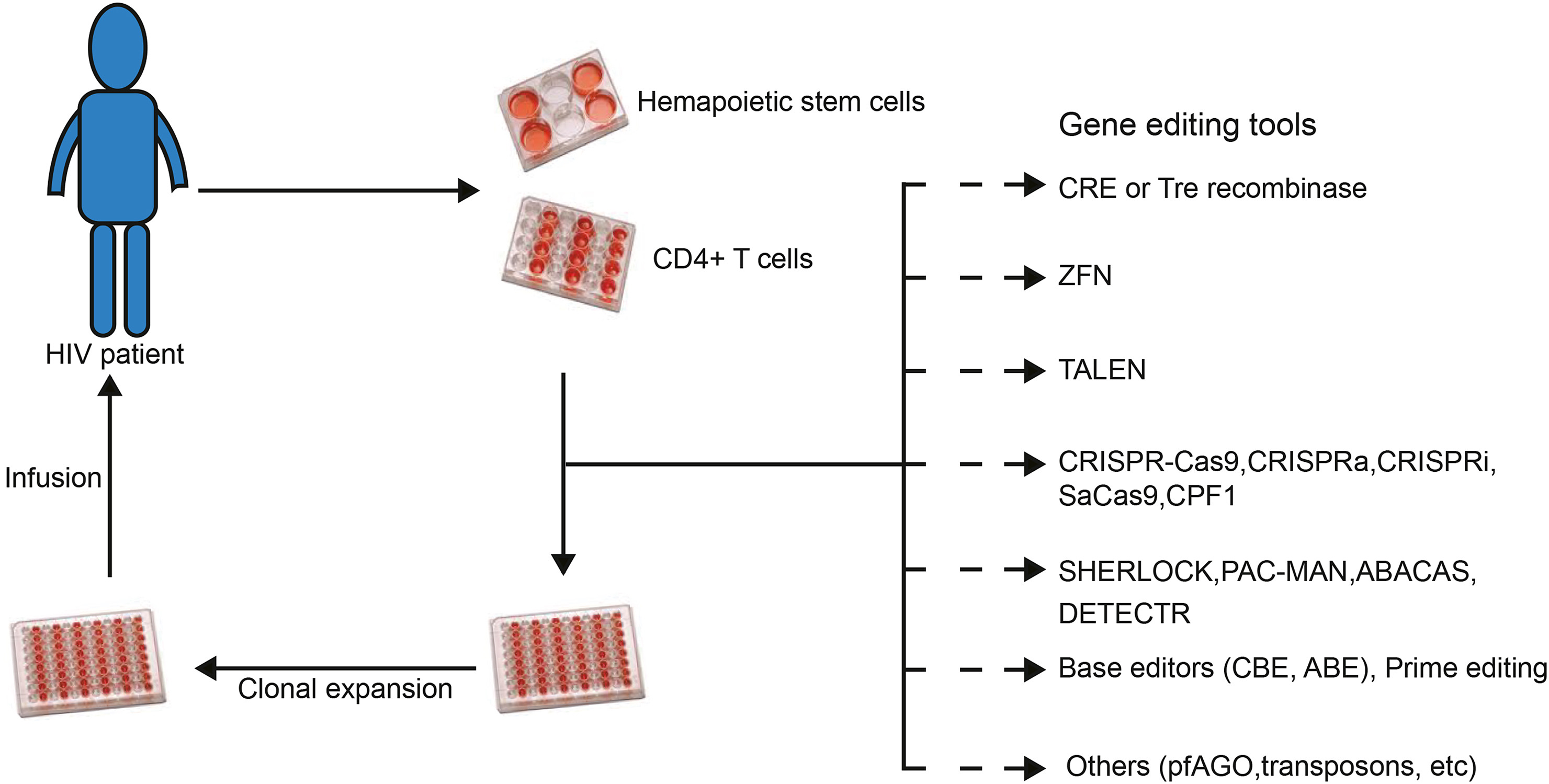
Zhihao Zhang, Wei Hou and Shuliang Chen. Updates on CRISPR-based gene editing in HIV-1/AIDS therapy[J]. Virologica Sinica, 2022, 37(1): 1-10. doi: 10.1016/j.virs.2022.01.017.
Although tremendous efforts have been made to prevent and treat HIV-1 infection, HIV-1/AIDS remains a major threat to global human health. The combination antiretroviral therapy (cART), although able to suppress HIV-1 replication, cannot eliminate the proviral DNA integrated into the human genome and thus requires lifelong treatment that may lead to various side effects. In recent years, clustered regularly interspaced short palindromic repeat (CRISPR)-associated nuclease 9 (Cas9) related gene-editing systems have been developed and designed as effective ways to treat HIV-1 infection. However, new gene-targeting tools derived from or functioning like CRISPR/Cas9, including base editor, prime editing, SHERLOCK, DETECTR, PAC-MAN, ABACAS, pfAGO, have been developed and optimized for pathogens detection and diseases correction. Here, we summarize recent studies on HIV-1/AIDS gene therapy and provide more gene-editing targets based on studies relating to the molecular mechanism of HIV-1 infection. We also identify the strategies and potential applications of these new gene-editing technologies for HIV-1/AIDS treatment in the future. Moreover, we discuss the caveats and problems that should be addressed before the clinical use of these versatile CRISPR-based gene targeting tools. Finally, we offer alternative solutions to improve the practice of gene targeting in HIV-1/AIDS gene therapy.
Jialu Zheng, Yutong Wei and Guan-Zhu Han. The diversity and evolution of retroviruses: Perspectives from viral “fossils”[J]. Virologica Sinica, 2022, 37(1): 11-18. doi: 10.1016/j.virs.2022.01.019.
Retroviruses exclusively infect vertebrates, causing a variety of diseases. The replication of retroviruses requires reverse transcription and integration into host genomes. When infecting germline cells, retroviruses become inherited vertically, forming endogenous retroviruses (ERVs). ERVs document past viral infections, providing molecular fossils for studying the evolutionary history of retroviruses. In this review, we summarize the recent advances in understanding the diversity and evolution of retroviruses from the perspectives of viral fossils, and discuss the effects of ERVs on the evolution of host biology.
Yingshuo Ma, Man Li, Lyu Xie, Na Gao, Dongying Fan, Kaihao Feng, Yao Yao, Yong Zhou, Ziyang Sheng, Hongning Zhou, Hui Chen and Jing An. Seroepidemiologic study on convalescent sera from dengue fever patients in Jinghong, Yunnan[J]. Virologica Sinica, 2022, 37(1): 19-29. doi: 10.1016/j.virs.2021.12.001.
After dengue virus (DENV) infection, antibody-dependent enhancement (ADE) is easy to occur when the neutralizing antibody (NAb) gradually decreases to a sub-neutralizing concentration. In this cohort surveillance, we utilized sera samples collected from dengue fever patients at different convalescent phases in Jinghong City, to investigate the dynamic change rule of DENV-specific antibodies, and to analyze the risk of ADE caused by secondary infection with heterologous serotypes DENVs. For baseline serosurvey, 191 four-year and 99 six-year sera samples during convalescence were collected in 2017 and 2019, respectively. The positive rate of DENV-specific immunoglobulin G was 98.4% in 2017, which significantly decreased to 82.8% in 2019. The geometric mean titer (GMT) of NAb decreased from 1:155.35 to 1:46.66. Among 290 overall samples, 73 paired consecutive samples were used for follow-up serosurvey. In four-year sera, the GMTs of NAb against DENV-3 and cross-reactive antibodies against DENV-1, DENV-2 and DENV-4 were 1:167.70, 1:13.80, 1:18.54 and 1:45.26, respectively, which decreased to 1:53.18, 1:10.30, 1:14.60 and 1:8.17 in six-year sera. In age-stratified analysis, due to the increasing number of ADE positive samples from 2017 to 2019 in 31–40 and 51–60 years groups, the risk of ADE in DENV-4 infection was positively associated with the extension of convalescent phase, and the odd ratio was higher than other groups. With the recovery period lengthened, the risk of secondary infection with DENV-1 and DENV-2 was reduced. Our results offer essential experimental data for risk prediction of severe dengue in hyper-endemic dengue areas, and provide crucial scientific insight for the development of effective dengue vaccines.
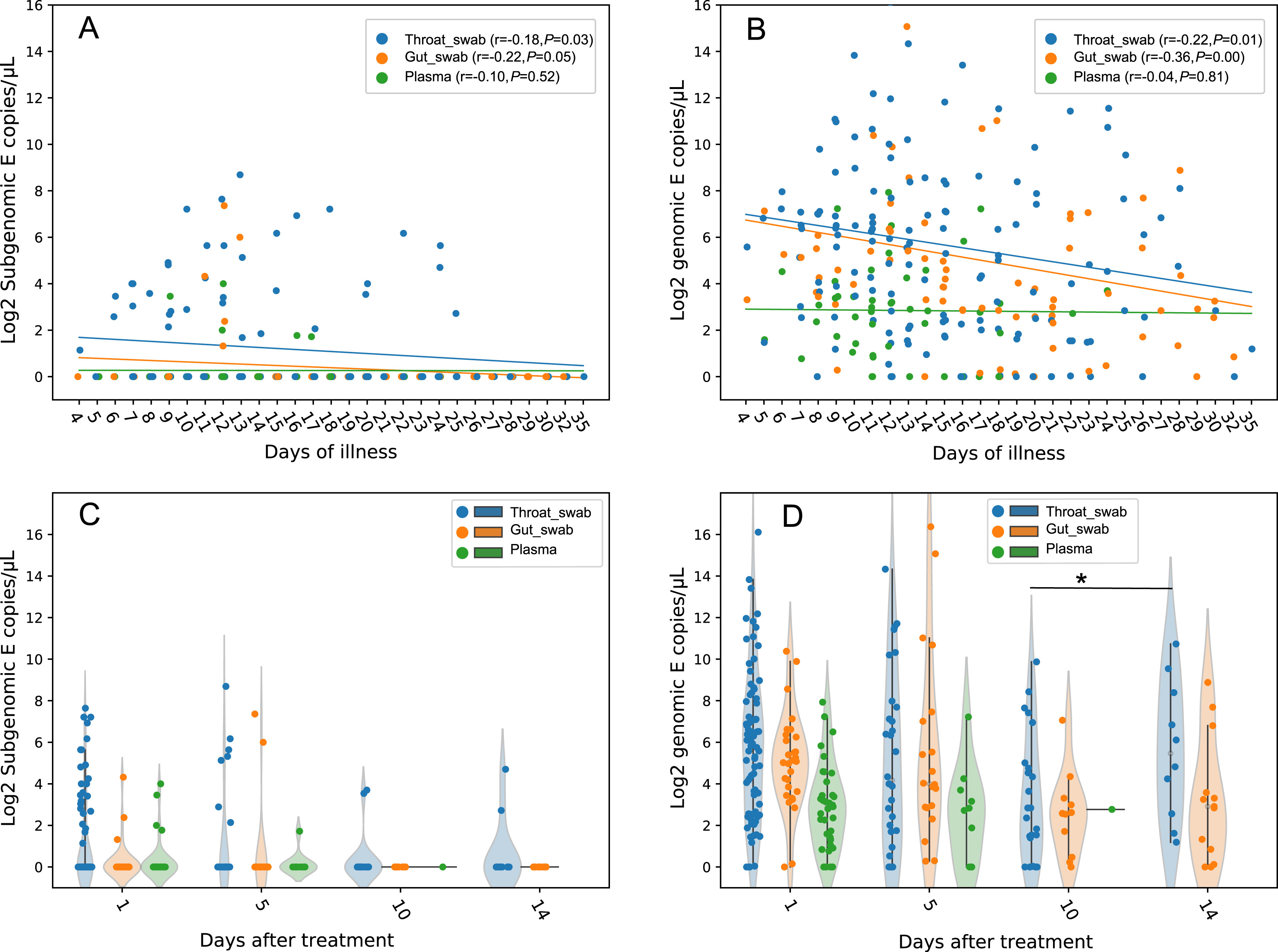
Xiaohui Zou, Shengrui Mu, Yeming Wang, Li Guo, Lili Ren, Xiaoyan Deng, Haibo Li, Jiankang Zhao, Yulin Zhang, Hui Li, Binghuai Lu, Chaolin Huang and Bin Cao. Characterization of two SARS-CoV-2 subgenomic RNA dynamics in severe COVID-19 patients[J]. Virologica Sinica, 2022, 37(1): 30-37. doi: 10.1016/j.virs.2022.01.008.
Little is known about Subgenomic RNA (sgRNA) dynamics in patients with Coronavirus diseases 2019 (COVID-19). We collected 147 throat swabs, 74 gut swabs and 46 plasma samples from 117 COVID-19 patients recruited in the LOTUS China trial (ChiCTR2000029308) and compared E and orf7a sgRNA load in patients with different illness duration, outcome, and comorbidities. Both sgRNAs were detected in all the three types of samples, with longest duration of 25, 13, and 17 days for E sgRNA, and 32, 28, and 17 days for orf7a sgRNA in throat, gut, and plasma, respectively. A total of 95% (57/60) of patients had no E sgRNA detected after 10 days post treatment, though 86% of them were still E RNA positive. High correlation on titer was observed between sgRNA encoding E and orf7a gene. sgRNA showed similar variation in the standard care and Lopinavir-Ritonavir group. Patients with diabetes and heart diseases showed higher pharyngeal E sgRNA at the first day (P = 0.016 and 0.013, respectively) but no difference at five days after treatment, compared with patients without such commodities. Patients with hypertension and cerebrovascular diseases showed no difference in the pharyngeal sgRNA levels at both one and five days after treatment, compared with patients without these two commodities. E sgRNA levels in the initial infection showed no correlation with the serum antibody against spike, nucleoprotein, and receptor binding domains at ten days later. sgRNA lasted a long period in COVID-19 patients and might have little effect on humoral response.
Aobaixue Zhou, Jiahao Zhang, Huanan Li, Qiang Xu, Yiqun Chen, Bo Li, Wanying Liu, Guanming Su, Xingxing Ren, Guangjie Lao, Baozheng Luo, Ming Liao and Wenbao Qi. Combined insertion of basic and non-basic amino acids at hemagglutinin cleavage site of highly pathogenic H7N9 virus promotes replication and pathogenicity in chickens and mice[J]. Virologica Sinica, 2022, 37(1): 38-47. doi: 10.1016/j.virs.2022.01.001.
Since mid-2016, the low pathogenic H7N9 influenza virus has evolved into a highly pathogenic (HP) phenotype in China, raising many concerns about public health and poultry industry. The insertion of a “KRTA” motif at hemagglutinin cleavage site (HACS) occurred in the early stage of HP H7N9 variants. During the co-circulation, the HACS of HP-H7N9 variants were more polymorphic in birds and humans. Although HP-H7N9 variants, unlike the H5 subtype virus, exhibited the insertions of basic and non-basic amino acids, the underlying function of those insertions and substitutions remains unclear. The results of bioinformatics analysis indicated that the PEVPKRKRTAR/G motif of HACS had become the dominant motif in China. Then, we generated six H7N9 viruses bearing the PEIPKGR/G, PEVPKGR/G, PEVPKRKRTAR/G, PEVPKGKRTAR/G, PEVPKGKRIAR/G, and PEVPKRKRR/G motifs. Interestingly, after the deletion of threonine and alanine (TA) at HACS, the H7N9 viruses manifested decreased thermostability and virulence in mice, and the PEVPKRKRTAR/G-motif virus is prevalent in birds and humans probably due to its increased transmissibility and moderate virulence. By contrast, the insertion of non-basic amino acid isoleucine and alanine (IA) decreased the transmissibility in chickens and virulence in mice. Remarkably, the I335V substitution of H7N9 virus enhanced infectivity and transmission in chickens, suggesting that the combination of mutations and insertions of amino acids at the HACS promoted replication and pathogenicity in chickens and mice. The ongoing evolution of H7N9 increasingly threatens public health and poultry industry, so, its comprehensive surveillance and prevention of H7N9 viruses should be pursued.
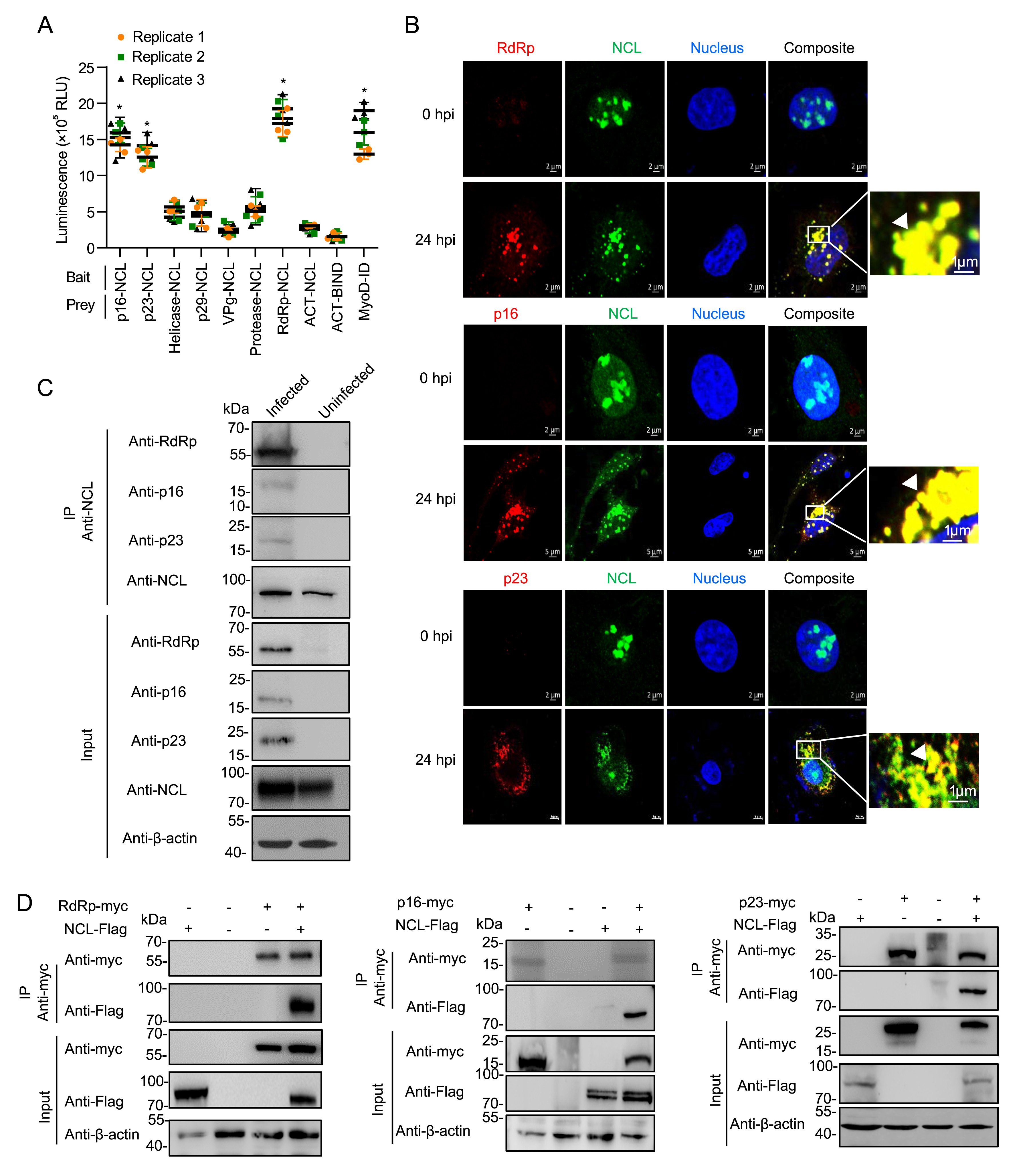
Jie Zhu, Qiuhong Miao, Hongyuan Guo, Aoxing Tang, Dandan Dong, Jingyu Tang, Fang Wang, Guangzhi Tong and Guangqing Liu. Nucleolin interacts with the rabbit hemorrhagic disease virus replicase RdRp, nonstructural proteins p16 and p23, playing a role in virus replication[J]. Virologica Sinica, 2022, 37(1): 48-59. doi: 10.1016/j.virs.2022.01.004.
Rabbit hemorrhagic disease virus (RHDV) is a member of the Caliciviridae family and cannot be propagated in vitro, which has impeded the progress of investigating its replication mechanism. Construction of an RHDV replicon system has recently provided a platform for exploring RHDV replication in host cells. Here, aided by this replicon system and using two-step affinity purification, we purified the RHDV replicase and identified its associated host factors. We identified rabbit nucleolin (NCL) as a physical link, which mediating the interaction between other RNA-dependent RNA polymerase (RdRp)-related host proteins and the viral replicase RdRp. We found that the overexpression or knockdown of NCL significantly increased or severely impaired RHDV replication in RK-13 cells, respectively. NCL was identified to directly interact with RHDV RdRp, p16, and p23. Furthermore, NCL knockdown severely impaired the binding of RdRp to RdRp-related host factors. Collectively, these results indicate that the host protein NCL is essential for RHDV replication and acts as a physical link between viral replicase and host proteins.
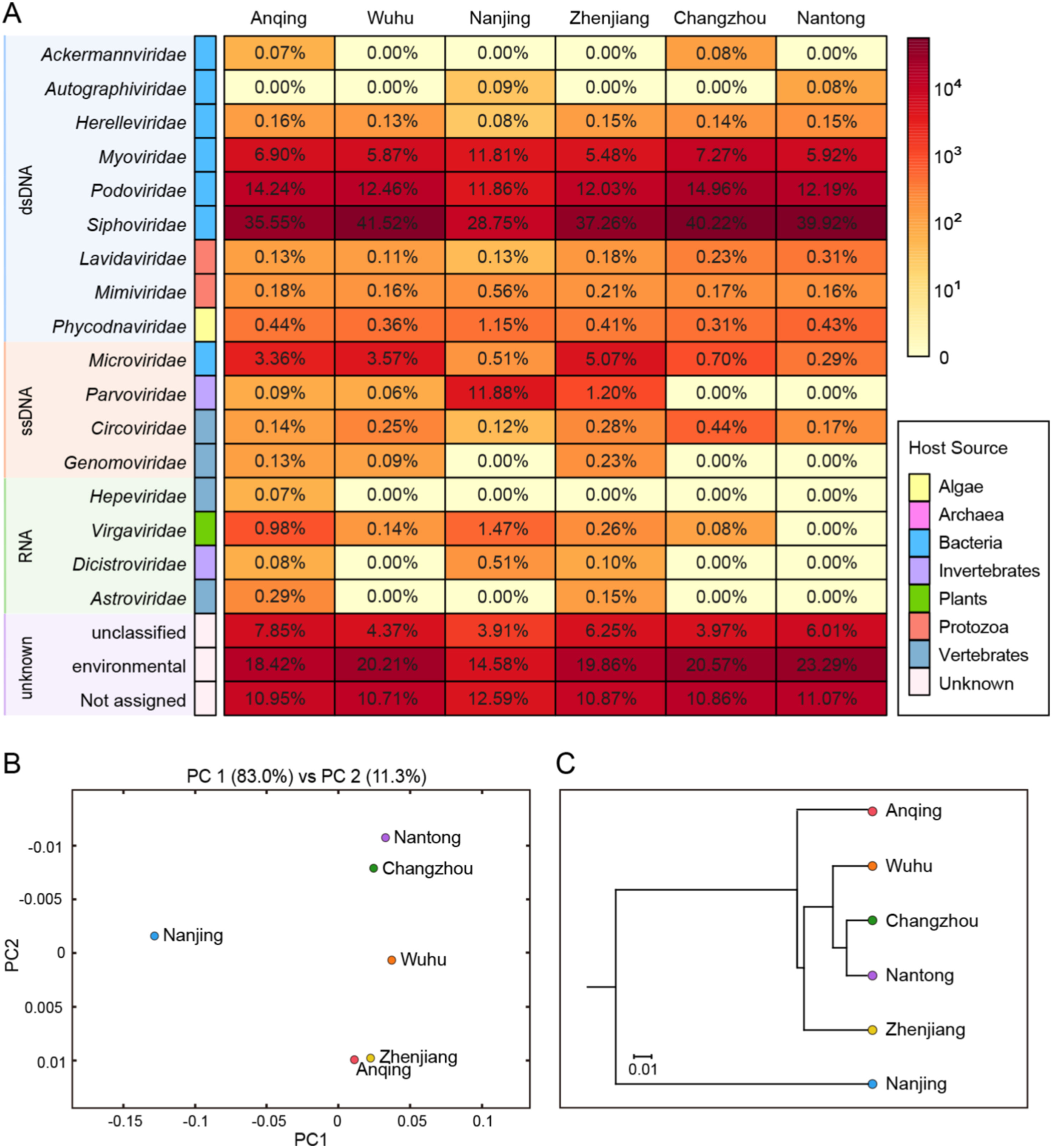
Juan Lu, Shixing Yang, Xiaodan Zhang, Xiangming Tang, Ju Zhang, Xiaochun Wang, Hao Wang, Quan Shen and Wen Zhang. Metagenomic analysis of viral community in the Yangtze River expands known eukaryotic and prokaryotic virus diversity in freshwater[J]. Virologica Sinica, 2022, 37(1): 60-69. doi: 10.1016/j.virs.2022.01.003.
Viruses in aquatic ecosystems are characterized by extraordinary abundance and diversity. Thus far, there have been limited studies focused on viral communities in river water systems. Here, we investigated the virome of the Yangtze River Delta using viral metagenomic analysis. The compositions of viral communities from six sampling sites were analyzed and compared. By using library construction and next generation sequencing, contigs and singlet reads similar to viral sequences were classified into 17 viral families, including nine dsDNA viral families, four ssDNA viral families and four RNA viral families. Statistical analysis using Friedman test suggested that there was no significant difference among the six sampling sites (P > 0.05). The viromes in this study were all dominated by the order Caudovirales, and a group of Freshwater phage uvFW species were particularly prevalent among all the samples. The virome from Nanjing presented a unique pattern of viral community composition with a relatively high abundance of family Parvoviridae. Phylogenetic analyses based on virus hallmark genes showed that the Caudovirales order and CRESS-DNA viruses presented high genetic diversity, while viruses in the Microviridae and Parvoviridae families and the Riboviria realm were relatively conservative. Our study provides the first insight into viral community composition in large river ecosystem, revealing the diversity and stability of river water virome, contributing to the proper utilization of freshwater resource.
Ouyang Peng, Xiaona Wei, Usama Ashraf, Fangyu Hu, Yongbo Xia, Qiuping Xu, Guangli Hu, Chunyi Xue, Yongchang Cao and Hao Zhang. Genome-wide transcriptome analysis of porcine epidemic diarrhea virus virulent or avirulent strain-infected porcine small intestinal epithelial cells[J]. Virologica Sinica, 2022, 37(1): 70-81. doi: 10.1016/j.virs.2022.01.011.
Porcine epidemic diarrhea virus (PEDV) is the main cause of diarrhea, vomiting, and mortality in pigs, which results in devastating economic loss to the pig industry around the globe. In recent years, the advent of RNA-sequencing technologies has led to delineate host responses at late stages of PEDV infection; however, the comparative analysis of host responses to early-stage infection of virulent and avirulent PEDV strains is currently unknown. Here, using the BGI DNBSEQ RNA-sequencing, we performed global gene expression profiles of pig intestinal epithelial cells infected with virulent (GDS01) or avirulent (HX) PEDV strains for 3, 6, and 12 h. It was observed that over half of all significantly dysregulated genes in both infection groups exhibited a down-regulated expression pattern. Functional enrichment analyses indicated that the differentially expressed genes (DEGs) in the GDS01 group were predominantly related to autophagy and apoptosis, whereas the genes showing the differential expression in the HX group were strongly enriched in immune responses/inflammation. Among the DEGs, the functional association of TLR3 and IFIT2 genes with the HX and GDS01 strains replication was experimentally validated by TLR3 inhibition and IFIT2 overexpression systems in cultured cells. TLR3 expression was found to inhibit HX strain, but not GDS01 strain, replication by enhancing the IFIT2 expression in infected cells. In conclusion, our study highlights similarities and differences in gene expression patterns and cellular processes/pathways altered at the early-stage infection of PEDV virulent and avirulent strains. These findings may provide a foundation for establishing novel therapies to control PEDV infection.
Shixing Yang, Yumin He, Ju Zhang, Dianqi Zhang, Yan Wang, Xiang Lu, Xiaochun Wang, Quan Shen, Likai Ji, Hongyan Lu and Wen Zhang. Viral metagenomics reveals diverse viruses in the fecal samples of children with diarrhea[J]. Virologica Sinica, 2022, 37(1): 82-93. doi: 10.1016/j.virs.2022.01.012.
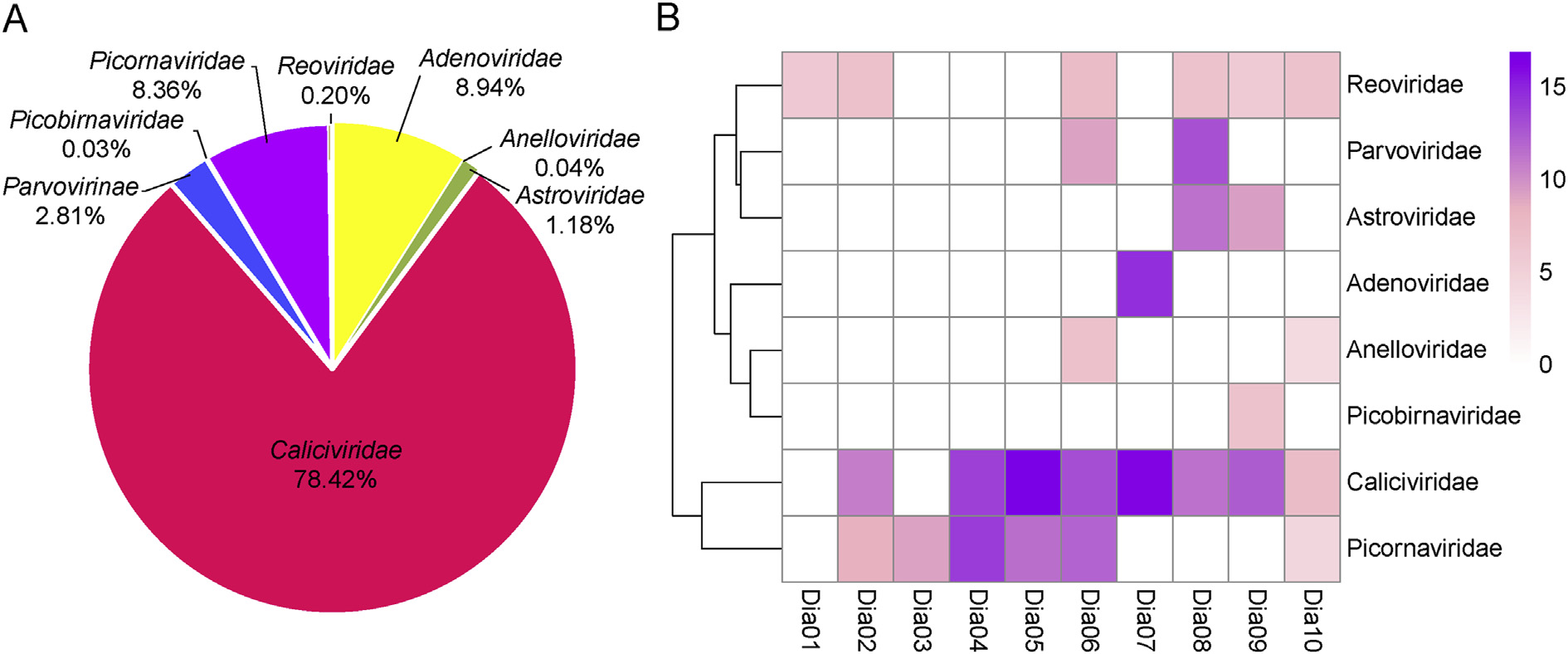
Diarrhea is the third leading cause of death in developing countries in children under the age of five. About half a million children die of diarrhea every year, most of which in developing countries. Viruses are the main pathogen of diarrhea. In China, the fecal virome of children with diarrhea has been rarely studied. Using an unbiased viral metagenomics approach, we analyzed the fecal virome in children with diarrhea. Many DNA or RNA viruses associated with diarrhea identified in those fecal samples were mainly from six families of Adenoviridae, Astroviridae, Caliciviridae, Parvoviridae, Picornaviridae, and Reoviridae. Among them, the family of Caliciviridae accounts for the largest proportion of 78.42%, following with Adenoviridae (8.94%) and Picornaviridae (8.36%). In addition to those diarrhea-related viruses that have already been confirmed to cause human diarrhea, the viruses not associated with diarrhea were also identified including anellovirus and picobirnavirus. This study increased our understanding of diarrheic children fecal virome and provided valuable information for the prevention and treatment of viral diarrhea in this area.
Muhammad Imran, Luping Zhang, Bohan Zheng, Zikai Zhao, Dengyuan Zhou, Shengfeng Wan, Zheng Chen, Hongyu Duan, Qiuyan Li, Xueqin Liu, Shengbo Cao, Shaoyong Ke and Jing Ye. Screening of novel synthetic derivatives of dehydroepiandrosterone for antivirals against flaviviruses infections[J]. Virologica Sinica, 2022, 37(1): 94-106. doi: 10.1016/j.virs.2022.01.007.
Flaviviruses are important arthropod-borne pathogens that represent an immense global health problem. Their unprecedented epidemic rate and unpredictable clinical features underscore an urgent need for antiviral interventions. Dehydroepiandrosterone (DHEA) is a natural occurring adrenal-derived steroid in the human body that has been associated in protection against various infections. In the present study, the plaque assay based primary screening was conducted on 32 synthetic derivatives of DHEA against Japanese encephalitis virus (JEV) to identify potent anti-flaviviral compounds. Based on primary screening, HAAS-AV3026 and HAAS-AV3027 were selected as hits from DHEA derivatives that exhibited strong antiviral activity against JEV (IC50 = 2.13 and 1.98 μmol/L, respectively) and Zika virus (ZIKV) (IC50 = 3.73 and 3.42 μmol/L, respectively). Mechanism study indicates that HAAS-AV3026 and HAAS-AV3027 do not exhibit inhibitory effect on flavivirus binding and entry process, while significantly inhibit flavivirus infection at the replication stage. Moreover, indirect immunofluorescence assay, Western blot analyses, and quantitative reverse transcription-PCR (qRT-PCR) revealed a potent antiviral activity of DHEA derivatives hits against JEV and ZIKV in terms of inhibition of viral infection, protein production, and viral RNA synthesis in Vero cells. Taken together, our results may provide a basis for the development of new antivirals against flaviviruses.
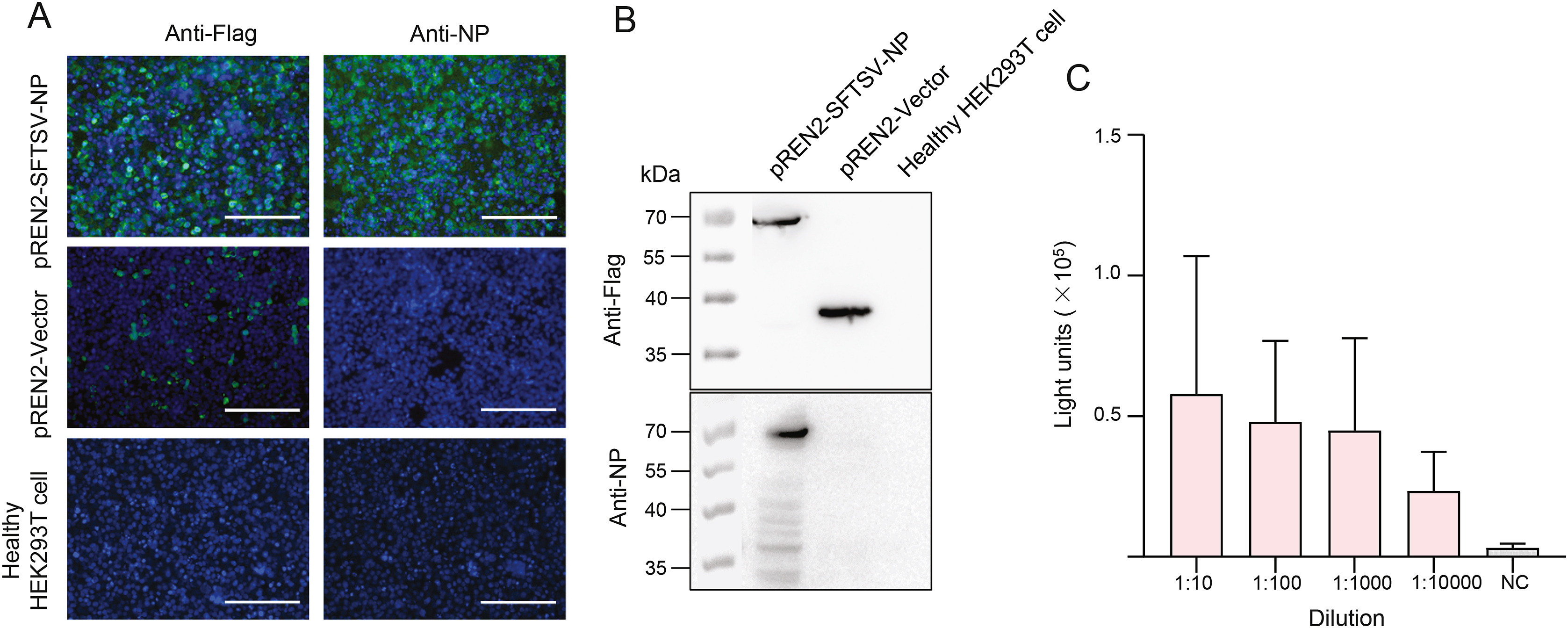
Shengyao Chen, Minjun Xu, Xiaoli Wu, Yuan Bai, Junming Shi, Min Zhou, Qiaoli Wu, Shuang Tang, Fei Deng, Bo Qin and Shu Shen. A new luciferase immunoprecipitation system assay provided serological evidence for missed diagnosis of severe fever with thrombocytopenia syndrome[J]. Virologica Sinica, 2022, 37(1): 107-114. doi: 10.1016/j.virs.2022.01.018.
Severe fever with thrombocytopenia syndrome (SFTS), caused by SFTS virus (SFTSV) infection, was first reported in 2010 in China with an initial fatality of up to 30%. The laboratory confirmation of SFTSV infection in terms of detection of viral RNA or antibody levels is critical for SFTS diagnosis and therapy. In this study, a new luciferase immunoprecipitation system (LIPS) assay based on pREN2 plasmid expressing SFTSV NP gene and tagged with Renilla luciferase (Rluc), was established and used to investigate the levels of antibody responses to SFTSV. Totally 464 serum samples from febrile patients were collected in the hospital of Shaoxing City in Zhejiang Province in 2019. The results showed that 82 of the 464 patients (17.7%) had antibody response to SFTSV, which were further supported by immunofluorescence assays (IFAs). Further, qRT-PCR and microneutralization tests showed that among the 82 positive cases, 15 patients had viremia, 10 patients had neutralizing antibody, and one had both (totally 26 patient). However, none of these patients were diagnosed as SFTS in the hospital probably because of their mild symptoms or subclinical manifestations. All the results indicated that at least the 26 patients having viremia or neutralizing antibody were the missed diagnosis of SFTS cases. The findings suggested the occurrence of SFTS and the SFTS incidence were higher than the reported level in Shaoxing in 2019, and that LIPS may provide an alternative strategy to confirm SFTSV infection in the laboratory.
Zhihua Liu, Yawei Zhang, Mengli Cheng, Ningning Ge, Jiayi Shu, Zhiheng Xu, Xiao Su, Zhihua Kou, Yigang Tong, Chengfeng Qin and Xia Jin. A single nonsynonymous mutation on ZIKV E protein-coding sequences leads to markedly increased neurovirulence in vivo[J]. Virologica Sinica, 2022, 37(1): 115-126. doi: 10.1016/j.virs.2022.01.021.
Zika virus (ZIKV) can infect a wide range of tissues including the developmental brain of human fetus. Whether specific viral genetic variants are linked to neuropathology is incompletely understood. To address this, we have intracranially serially passaged a clinical ZIKV isolate (SW01) in neonatal mice and discovered variants that exhibit markedly increased virulence and neurotropism. Deep sequencing analysis combining with molecular virology studies revealed that a single 67D (Aspartic acid) to N (Asparagine) substitution on E protein is sufficient to confer the increased virulence and neurotropism in vivo. Notably, virus clones with D67N mutation had higher viral production and caused more severe cytopathic effect (CPE) in human neural astrocytes U251 cells in vitro, indicating its potential neurological toxicity to human brain. These findings revealed that a single mutation D67N on ZIKV envelope may lead to severe neuro lesion that may help to explain the neurovirulence of ZIKV and suggest monitoring the occurrence of this mutation during nature infection may be important.
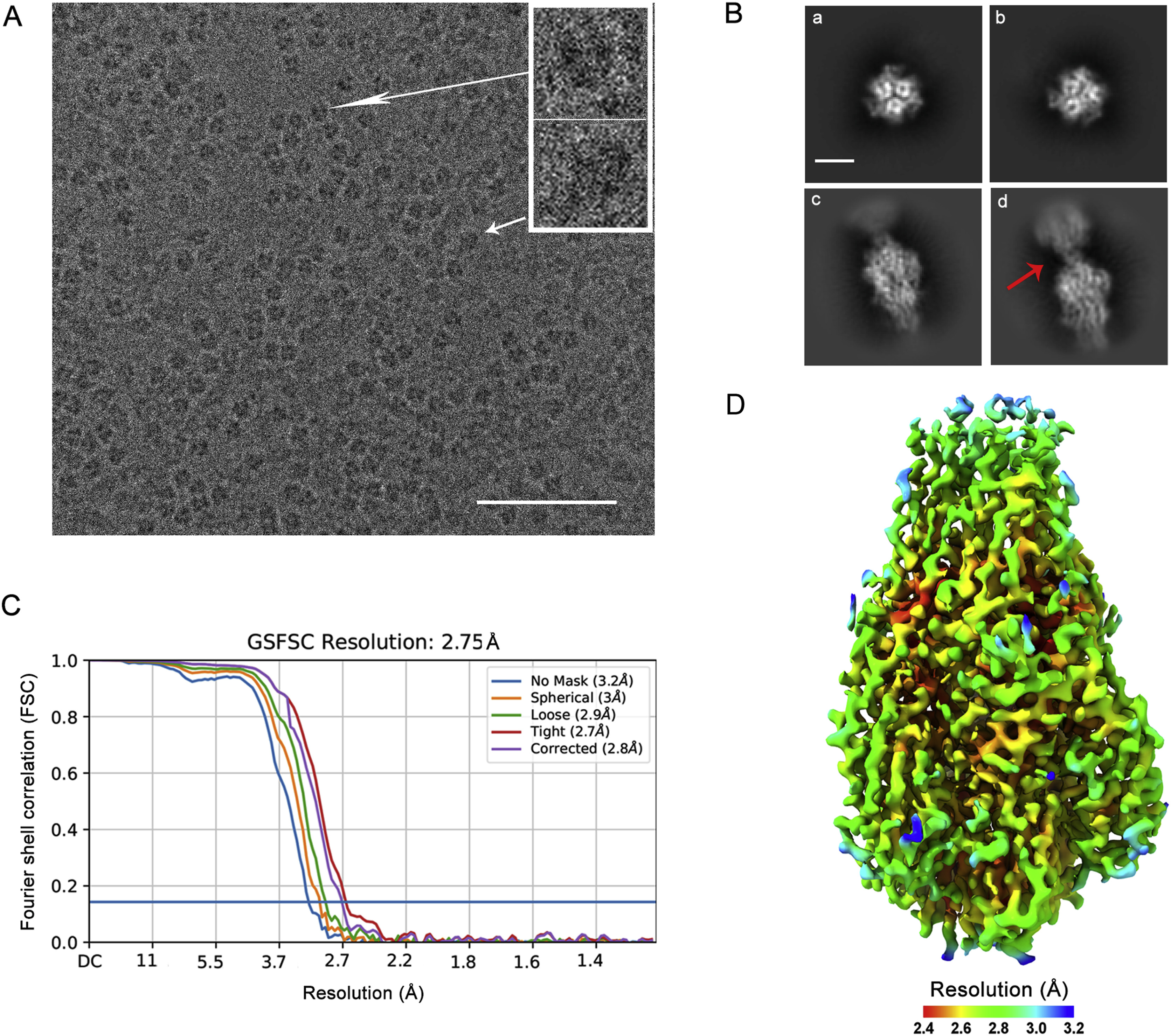
Na Li, Guibo Rao, Zhiqiang Li, Jiayi Yin, Tingting Chong, Kexing Tian, Yan Fu and Sheng Cao. Cryo-EM structure of glycoprotein C from Crimean-Congo hemorrhagic fever virus[J]. Virologica Sinica, 2022, 37(1): 127-137. doi: 10.1016/j.virs.2022.01.015.
Crimean-Congo hemorrhagic fever virus (CCHFV) is a causative agent of serious hemorrhagic diseases in humans with high mortality rates. CCHFV glycoprotein Gc plays critical roles in mediating virus-host membrane fusion and has been studied extensively as an immunogen. However, the molecular mechanisms involved in membrane fusion and Gc-specific antibody-antigen interactions remain unresolved largely because structural information of this glycoprotein is missing. We designed a trimeric protein including most of the ectodomain region of Gc from the prototype CCHFV strain, IbAr10200, which enabled the cryo-electron microscopy structure to be solved at a resolution of 2.8 Å. The structure confirms that CCHFV Gc is a class II fusion protein. Unexpectedly, structural comparisons with other solved Gc trimers in the postfusion conformation revealed that CCHFV Gc adopted hybrid architectural features of the fusion loops from hantaviruses and domain III from phenuiviruses, suggesting a complex evolutionary pathway among these bunyaviruses. Antigenic sites on CCHFV Gc that protective neutralizing antibodies target were mapped onto the CCHFV Gc structure, providing valuable information that improved our understanding of potential neutralization mechanisms of various antibodies.
Ying Tang, Yiqin Wang, Yuchang Li, Huai Zhao, Sen Zhang, Ying Zhang, Jing Li, Yuehong Chen, Xiaoyan Wu, Chengfeng Qin, Tao Jiang and Xiaoping Kang. An integrated rapid nucleic acid detection assay based on recombinant polymerase amplification for SARS-CoV-2[J]. Virologica Sinica, 2022, 37(1): 138-141. doi: 10.1016/j.virs.2022.01.006.
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is a novel coronavirus that causes the outbreak of coronavirus disease 2019 (COVID-19) (Li et al., 2020a). Viral nucleic acid testing is the standard method for the laboratory diagnosis of COVID-19 (Wu et al., 2020a; Zhu et al., 2020). Currently, a variety of qPCR-based detection kits are used for laboratory-based detection and confirmation of SARS-CoV-2 infection (Corman et al., 2020; Hussein et al., 2020; Ruhan et al., 2020; Veyer et al., 2020). Conventional qPCR involves virus inactivation, nucleic acid extraction, and qPCR amplification procedures. Therefore, the process is complicated, which usually takes longer than 2 h, and requires biosafety laboratories and professional staff. Thus, qPCR is not suitable for use in field or medical units. To reduce the operation steps, automatic integrated qPCR detection systems that combine nucleic acid extraction and qPCR amplification in a sealed cartridge were developed to detect viruses in clinical samples (Li et al., 2020b). However, the detection time is still longer than 1 h. Therefore, rapid nucleic acid detection systems are needed to further improve the detection efficiency.
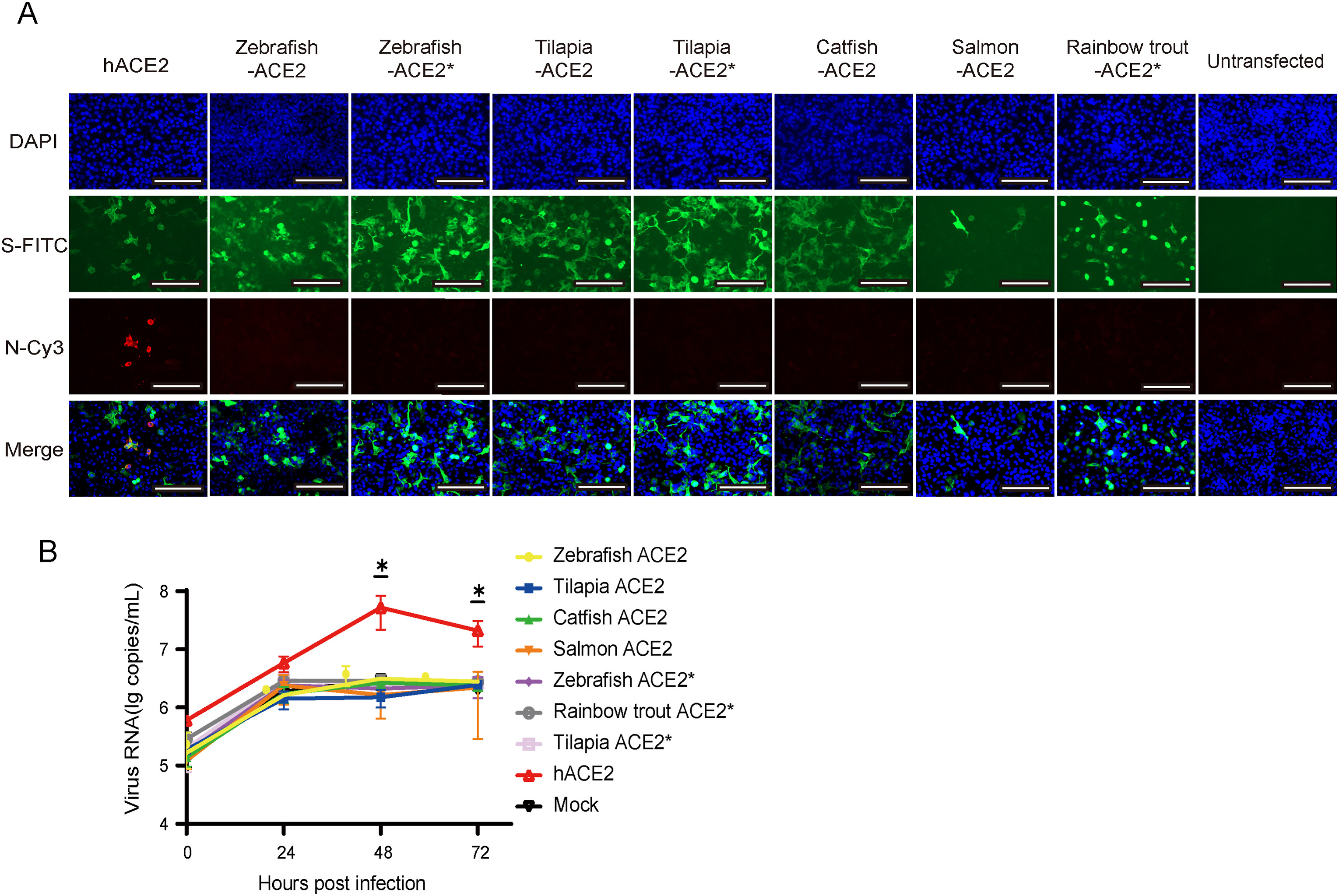
Shi-Zhe Xie, Mei-Qin Liu, Ren-Di Jiang, Hao-Feng Lin, Wei Zhang, Bei Li, Jia Su, Fei Ke, Qi-Ya Zhang, Zheng-Li Shi and Xing-Lou Yang. Fish ACE2 is not susceptible to SARS-CoV-2[J]. Virologica Sinica, 2022, 37(1): 142-144. doi: 10.1016/j.virs.2022.01.020.
The ongoing pandemic of coronavirus disease 2019 (COVID-19) has reshaped our daily life and caused > 4 million deaths worldwide (https://covid19.who.int/). Although lockdown and vaccination have improved the situation in many countries, imported cases and sporadic outbreaks pose a constant stress to the prevention and control of COVID-19. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the etiologic agent responsible for COVID-19, has a positive-sense single-stranded RNA genome of 30 kb (Coronaviridae Study Group of the International Committee on Taxonomy of Viruses, 2020). We and other groups have demonstrated that the SARS-CoV-2 could use the angiotensin-converting enzyme 2 (ACE2) as cell receptor, including orthologs of a broad range of animal species such as human, bats, ferrets, pigs, cats, and dogs (Hoffmann et al., 2020; Zhou et al., 2020; Liu et al., 2021). Although the evolutionary origin of SARS-CoV-2 can be linked to the discoveries of diverse coronaviruses related to SARS-CoV-2 in wild animals such as bats (Zhou et al., 2020; Wacharapluesadee et al., 2021) and pangolins (Liu et al., 2019; Lam et al., 2020), the direct origin of SARS-CoV-2 in humans remains unknown. In China, several sporadic outbreaks of COVID-19 in 2020 were linked to food in cold chain sold at trade markets, including salmon meat (http://www.nhc.gov.cn/xcs/yqtb/list_gzbd.shtml) (Yang et al., 2020). The detection of SARS-CoV-2 RNA on the surface of frozen meat for as long as 20 days has also been reported (Feng et al., 2021). A concern regarding the potential role of fish in SARS-CoV-2 transmission has also been raised. Therefore, we investigated the susceptibility of fish ACE2 to SARS-CoV-2.
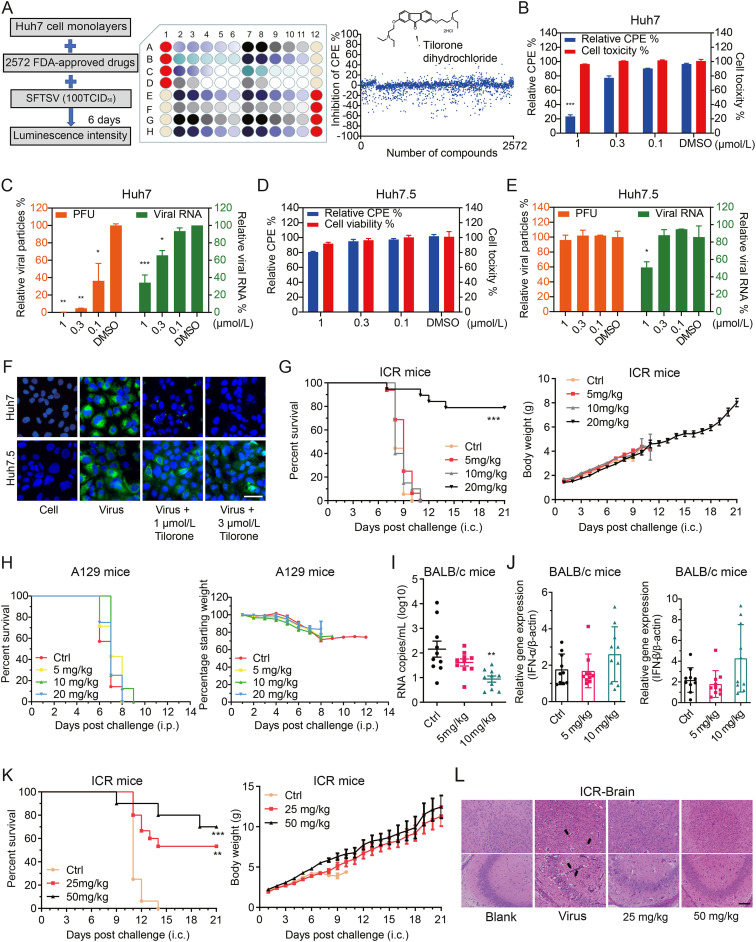
Jingjing Yang, Yunzheng Yan, Qingsong Dai, Jiye Yin, Lei Zhao, Yuexiang Li, Wei Li, Wu Zhong, Ruiyuan Cao and Song Li. Tilorone confers robust in vitro and in vivo antiviral effects against severe fever with thrombocytopenia syndrome virus[J]. Virologica Sinica, 2022, 37(1): 145-148. doi: 10.1016/j.virs.2022.01.014.
Severe fever with thrombocytopenia syndrome virus (SFTSV), an emerging pathogen, is a tick-borne bunyavirus belonging to the genus Bandavirus in the family Phenuiviridae (Kuhn et al., 2020). This pathogen was first identified in China during the heightened surveillance of acute febrile illness in 2009, and has been reported to cause several outbreaks in eastern Asia areas, including China, Japan, and Korea (Yu et al., 2011). Besides, Vietnam has also reported several confirmed SFTS cases (Tran et al., 2019). The mortality rate in hospitalised patients with SFTSV infection is up to 10%–30%. Moreover, SFTSV has been reported to possibly transmitted by the contact of body fluids from person-to-person, and extensive SFTSV contamination was detected in the patient rooms (Kim et al., 2015). These reports suggest that more stringent isolation measures are needed for the prevention of massive SFTSV outbreak.
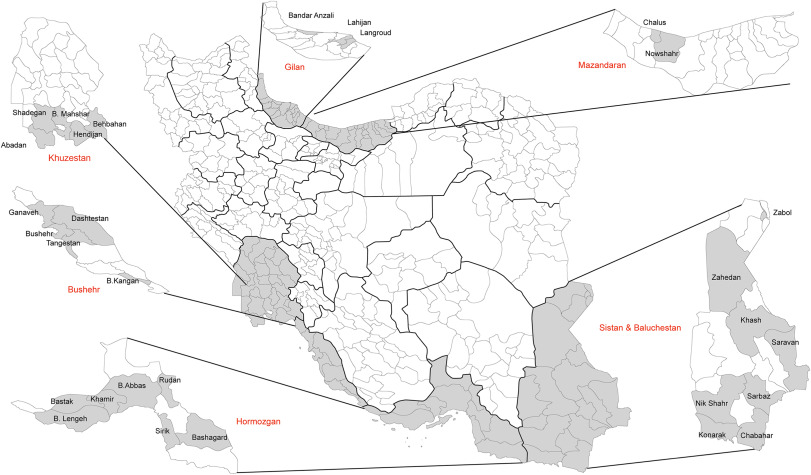
Abbas Ahmadi Vasmehjani, Farhad Rezaei, Mohammad Farahmand, Talat Mokhtari-Azad, Mohammad Reza Yaghoobi-Ershadi, Mohsen Keshavarz, Hamid Reza Baseri, Morteza Zaim, Mahmood Iranpour, Habibollah Turki and Mohammad Esmaeilpour-Bandboni. Epidemiological evidence of mosquito-borne viruses among persons and vectors in Iran: A study from North to South[J]. Virologica Sinica, 2022, 37(1): 149-152. doi: 10.1016/j.virs.2022.01.005.
Arthropod-borne viruses are a group of the most important emerging pathogens. They cause a range of diseases in vertebrate hosts and threaten human health (Gan and Leo, 2014). The global distribution of arboviruses is associated with the vector which is strongly affected by changes in environmental conditions. Dengue virus (DENV) and Chikungunya virus (CHIKV), which cause high annual infected cases and have an increasing geographic distribution, are transmitted by Aedes spp. mosquitoes, in particular Ae. albopictus and Ae. Aegypti (Presti et al., 2014; Higuera and Ramírez, 2018). Although, the main vector of dengue virus, Ae. aegypti, was not detected in Iran, other possible important vectors such as Ae. Albopictus and Ae. unilineatus were recorded (Doosti et al., 2016; Yaghoobi-Ershadi et al., 2017). West Nile virus (WNV), a member of the genus Flaviviruses, is one of the most widespread arboviruses (Chancey et al., 2015). The epidemiological evidence of WNV in different hosts in Iran was found (Bagheri et al., 2015), and the circulation of WNV in the main vector, Culex pipiens s.l. and Cx. pipiens, has been proved (Shahhosseini et al., 2017). Due to limited information on the situation of CHIKV, DENV and WNV in Iran, we performed a wide geographical investigation to determine the prevalence of IgG specific antibodies in human samples as well as the genome of WNV, CHIKV and DENV in mosquitoes.
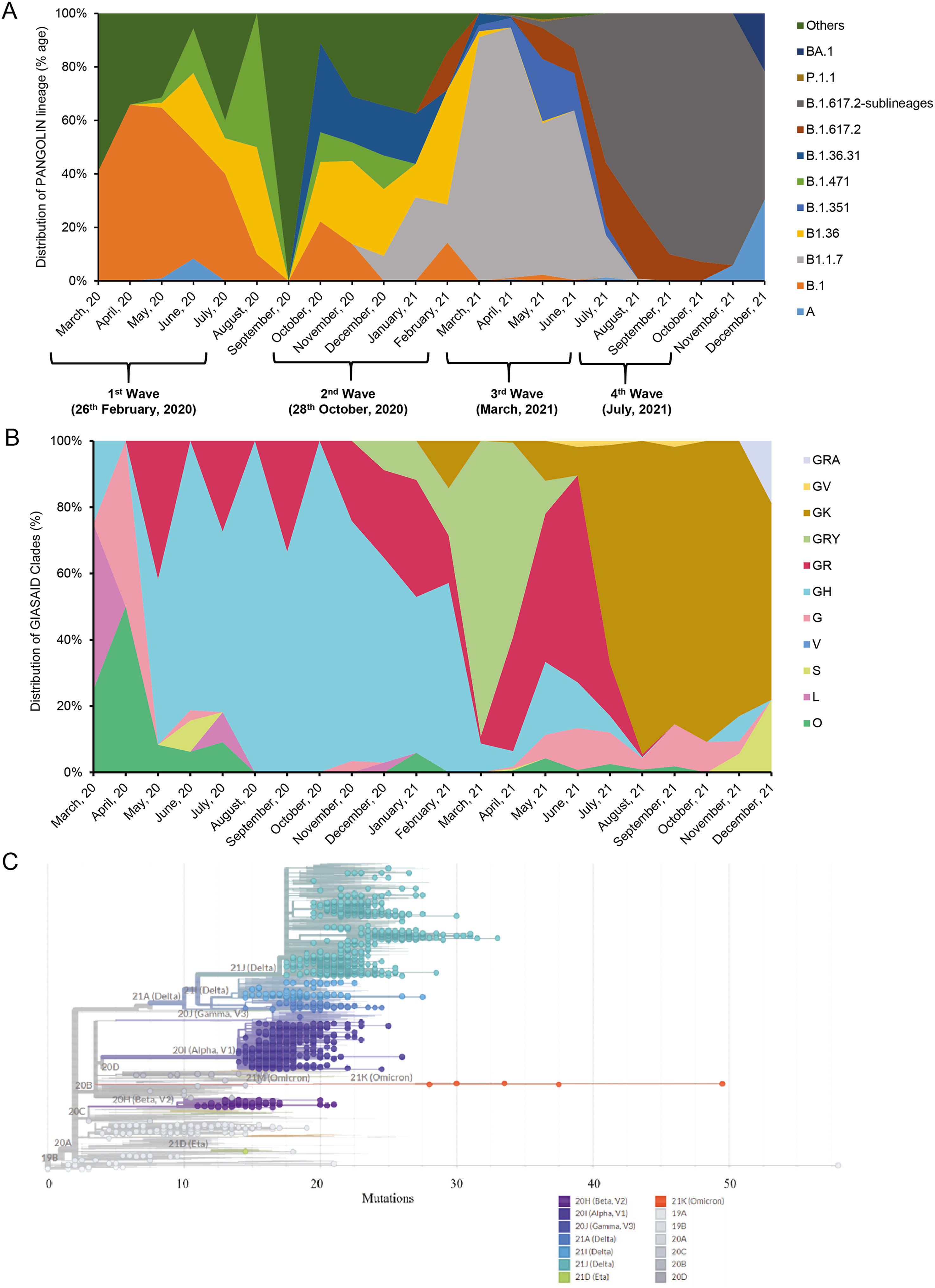
Zaira Rehman, Massab Umair, Aamer Ikram, Ammad Fahim and Muhammad Salman. Footprints of SARS-CoV-2 genome diversity in Pakistan, 2020–2021[J]. Virologica Sinica, 2022, 37(1): 153-155. doi: 10.1016/j.virs.2022.01.009.
The rapid spread of SARS-CoV-2 has significantly impacted the worldwide health system. The SARS-CoV-2 currently bears a remarkably low genetic diversity even though it carries one of the largest RNA genomes among viruses (Rausch et al., 2020). However, the coronaviruses harbor the capability of undergoing recombination at a high rate which can lead to the emergence of novel viral derivatives (Rausch et al., 2020; Gribble et al., 2021). This in turn requires not only global surveillance of SARS-CoV-2 genome in various countries but also careful scrutiny in animal genomic reservoirs. Conventionally, RNA viruses evolve with a high mutation rate, however, the presence of ExoN ribonuclease in SARS-CoV-2 genome has made its case different from other viral species (Gribble et al., 2021). The variables of natural selection which potentially drift the SARS-CoV-2 evolutionary dynamics can be recorded by analyzing deposited sequence genomes for its fitness, transmissibility potential, and pathogenicity (Rouchka et al., 2020). This can potentially provide a way to draw a holistic picture at a national level, while simultaneously providing a comparative overview with worldwide sequences.