Global HIV-1 sequence Patterns. The complexity of HIV-1 epidemiology in different areas of the world leaves unique footprints. Interestingly, the underlying significant patterns of global HIV-1 genomes could be detected using suitable data mining methods. Signature patterns of Env gene on characteristic amino acids are inferred from four HIV-1 pandemic regions and three primary subtypes worldwide. After cross-validation of these patterns, geographical and evolutionary associations were deduced. The significant pattern at position 21 indicates both the association of glycine, leucine and isoleucine with Africa and the Southern Africa, United States, Southeast Asia and China, and the association with subtypes C, B and CRF01_AE. It is expected that these patterns could provide a valuable reference for the epidemiological research or the design of vaccines in HIV studies.??Cover designed by Meng Wang, Wuhan IOV)
Crimean-Congo hemorrhagic fever (CCHF) is a zoonotic viral infection that is a serious threat to humans. The disease is widely distributed in Africa, Asia, and Europe and has developed into a serious public health concern. Humans become infected through the bites of ticks, by contact with a patient with CCHF, or by contact with blood or tissues from viremic livestock. Microvascular instability and impaired hemostasis are the hallmarks of the infection. Infection in human begins with nonspecific febrile symptoms, but may progress to a serious hemorrhagic syndrome with high mortality rates. Enzyme-linked immunoassay (ELISA) and polymerase chain reaction (PCR) are the most used and specific tests for the diagnosis. The mainstay of treatment is supportive. Although definitive studies are not available, ribavirin is suggested to be effective especially at the earlier phase of the infection. Uses of universal protective measures are the best way to avoid the infection. In this review, all aspects of CCHF are overviewed in light of the current literature.
Honghe Chen, Rongjuan Pei and Xinwen Chen. Different Responses of Two Highly Permissive Cell Lines Upon HCV Infection[J]. Virologica Sinica, 2013, 28(4): 202-208. doi: 10.1007/s12250-013-3342-5.
The construction of the first infectious clone JFH-1 speeds up the research on hepatitis C virus (HCV). However, Huh7 cell line was the only highly permissive cell line for HCV infection and only a few clones were fully permissive. In this study, two different fully permissive clones of Huh7 cells, Huh7.5.1 and Huh7-Lunet-CD81 (Lunet-CD81) cells were compared for their responses upon HCV infection. The virus replication level was found slightly higher in Huh7.5.1 cells than that in Lunet-CD81 cells. Viability of Huh7.5.1 cells but not of Lunet-CD81 cells was reduced significantly after HCV infection. Further analysis showed that the cell cycle of infected Huh7.5.1 cells was arrested at G1 phase. The G1/S transition was blocked by HCV infection in Huh7.5.1 cells as shown by the cell cycle synchronization analysis. Genes related to cell cycle regulation was modified by HCV infection and gene interaction analysis in GeneSpring GX in Direct Interactions mode highlighted 31 genes. In conclusion, the responses of those two cell lines were different upon HCV infection. HCV infection blocked G1/S transition and cell cycle progress, thus reduced the cell viability in Huh7.5.1 cells but not in Lunet-CD81 cells. Lunet-CD81 cells might be suitable for long term infection studies of HCV.
Hui Qiu, Fang Huang, Han Xiao, Binlian Sun and Rongge Yang. TRIM22 Inhibits the TRAF6-stimulated NF-κB Pathway by Targeting TAB2 for Degradation[J]. Virologica Sinica, 2013, 28(4): 209-215. doi: 10.1007/s12250-013-3343-4.
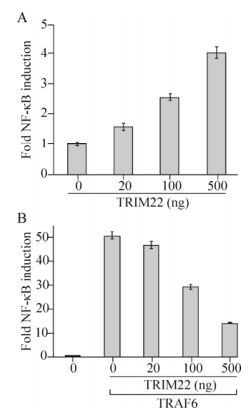
Tripartite motif containing 22 (TRIM22), a member of the TRIM/RBCC family, has been reported to activate the nuclear factor-kappa B (NF-κB) pathway in unstimulated macrophage cell lines, but the detailed mechanisms governing this activation remains unclear. We investigated this mechanism in HEK293T cells. We found that overexpression of TRIM22 could activate the NF-κB pathway and conversely, could inhibit the tumor necrosis factor receptor-associated factor 6 (TRAF6)-stimulated NF-κB pathway in HEK293T cells. Further experiments showed that TRIM22 could decrease the self-ubiquitination of TRAF6, and interact with and degrade transforming growth factor-β activated kinase 1 binding protein 2 (TAB2), and that these effects could be partially rescued by a TRIM22 RING domain deletion mutant. Collectively, our data indicate that overexpression of TRIM22 may negatively regulate the TRAF6-stimulated NF-κB pathway by interacting with and degrading TAB2.
Xiaohui Tian, Fei Zhao, Zhikui Cheng, Ming Zhou, Xiaoguang Zhi, Jiafu Li and Kanghong Hu. GCN5 Acetyltransferase Inhibits PGC1α-induced Hepatitis B Virus Biosynthesis[J]. Virologica Sinica, 2013, 28(4): 216-222. doi: 10.1007/s12250-013-3344-3.
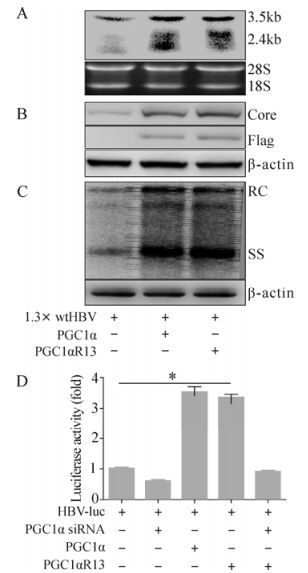
Hepatitis B virus (HBV) biosynthesis is primarily restricted to hepatocytes due to the governing of liver-enriched nuclear receptors (NRs) on viral RNA synthesis. The liver-enriched NR hepatocyte nuclear factor 4α (HNF4α), the key regulator of genes implicated in hepatic glucose metabolism, is also a primary determinant of HBV pregenomic RNA synthesis and HBV replication. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α) coactivates and further enhances the effect of HNF4α on HBV biosynthesis. Here, we showed that the acetyltransferase General Control Non-repressed Protein 5 (GCN5) acetylated PGC1α, leading to alteration of PGC1α from a transcriptionally active state into an inactive state. As a result, the coactivation activity of PGC1α on HBV transcription and replication was suppressed. Apparently, an acetylation site mutant of PGC1α (PGC1αR13) still had coactivation activity as GCN5 could not suppress the coactivation activity of the mutant. Moreover, a catalytically inactive acetyltransferase mutant GCN5m, due to the loss of acetylation activity, failed to inhibit the coactivation function of PGC1α in HBV biosynthesis. Our results demonstrate that GCN5, through its acetyltransferase activity, inhibits PGC1α-induced enhancement of HBV transcription and replication both in vitro and in vivo.
Pengcheng Yu, Xinjun Lv, Xinxin Shen, Qing Tang and Guodong Liang. Establishment and Preliminary Application of a Rapid Fluorescent Focus Inhibition Test (RFFIT) for Rabies Virus[J]. Virologica Sinica, 2013, 28(4): 223-227. doi: 10.1007/s12250-013-3321-x.
The World Health Organization (WHO) standard assay for determining levels of the rabies virus neutralization antibody (RVNA) is the rapid fluorescent focus inhibition test (RFFIT), which is used to evaluate the immunity effect after vaccination against rabies. For RFFIT, CVS-11 was used as the challenge virus, BSR cells as the adapted cells, and WHO rabies immunoglobulin (WHO STD) as the reference serum in this study. With reference to WHO and Pasteur RFFIT procedures, a micro-RFFIT procedure adapted to our laboratory was produced, and its specificity and reproducibility were tested. We tested levels of RVNA in human serum samples after immunization with different human rabies vaccines (domestic purified Vero cell rabies vaccine (PVRV) and imported purified chick embryo cell vaccine (PCECV)) using different regimens (Zagreb regimen and Essen regimen). We analyzed the levels of RVNA, and compared the immune efficacy of domestic PVRV and imported PCECV using different immunization regimens. The results showed that the immune efficacy of domestic PVRV using the Zagreb regimen was as good as that of the imported PCECV, but virus antibodies were generated more rapidly with the Zagreb regimen than with the Essen regimen. The RFFIT procedure established in our laboratory will enhance the comprehensive detection ability of institutions involved in rabies surveillance in China.
Yan Wang, Reda Rawi, Daniel Hoffmann, Binlian Sun and Rongge Yang. Inference of Global HIV-1 Sequence Patterns and Preliminary Feature Analysis[J]. Virologica Sinica, 2013, 28(4): 228-238. doi: 10.1007/s12250-013-3348-z.
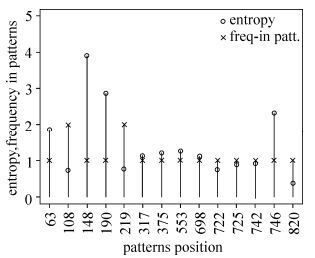
The epidemiology of HIV-1 varies in different areas of the world, and it is possible that this complexity may leave unique footprints in the viral genome. Thus, we attempted to find significant patterns in global HIV-1 genome sequences. By applying the rule inference algorithm RIPPER (Repeated Incremental Pruning to Produce Error Reduction) to multiple sequence alignments of Env sequences from four classes of compiled datasets, we generated four sets of signature patterns. We found that these patterns were able to distinguish southeastern Asian from non- southeastern Asian sequences with 97.5% accuracy, Chinese from non-Chinese sequences with 98.3% accuracy, African from non-African sequences with 88.4% accuracy, and southern African from non-southern African sequences with 91.2% accuracy. These patterns showed different associations with subtypes and with amino acid positions. In addition, some signature patterns were characteristic of the geographic area from which the sample was taken. Amino acid features corresponding to the phylogenetic clustering of HIV-1 sequences were consistent with some of the deduced patterns. Using a combination of patterns inferred from subtypes B, C, and all subtypes chimeric with CRF01_AE worldwide, we found that signature patterns of subtype C were extremely common in some sampled countries (for example, Zambia in southern Africa), which may hint at the origin of this HIV-1 subtype and the need to pay special attention to this area of Africa. Signature patterns of subtype B sequences were associated with different countries. Even more, there are distinct patterns at single position 21 with glycine, leucine and isoleucine corresponding to subtype C, B and all possible recombination forms chimeric with CRF01_AE, which also indicate distinct geographic features. Our method widens the scope of inference of signature from geographic, genetic, and genomic viewpoints. These findings may provide a valuable reference for epidemiological research or vaccine design.
Muaz M. Abdellatif, Bashir Salim, Awad A. Ibrahim, Tigani Asil and Abdelmalik I. Khalafalla. Analysis of TK and C18L Genes of Wild-type and Cell Culture Passaged Camelpox Virus[J]. Virologica Sinica, 2013, 28(4): 239-241. doi: 10.1007/s12250-013-3329-2.
Camelpox is an infectious skin disease of camels caused by camelpox virus (CMLV). It is confined to camel rearing belts of developing countries (Wernery U, et al., 2002). CMLV is classified in the Orthopoxvirus (OPV) genus of the subfamily Chordopoxvirinae in the Poxviridae family.
Yi Hu and Jingxin Zhang. Screening Strategies to Identify Host Factors Associated with the Early and Late Stages of HIV-1 Life Cycle[J]. Virologica Sinica, 2013, 28(4): 242-245. doi: 10.1007/s12250-013-3359-9.
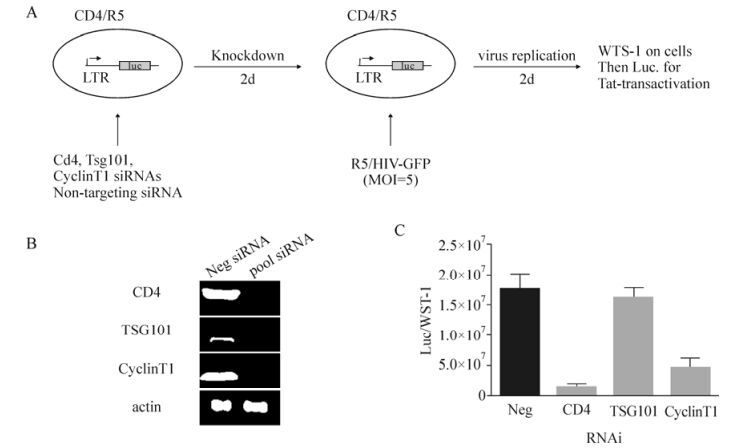
The HIV-1 infection in humans has now become a pandemic and in the absence of treatment it often leads to acquired immunodeficiency syndrome (AIDS). Although significant progress has been made to reduce the HIV-1 viral load in the blood of infected patients, challenges still exist in areas such as viral latency and viral resistance (Hosseinipour M C, 2013; Katlama C, 2013).