2021 Vol.36(1)
Hand, foot and mouth disease (HFMD), an infectious disease mostly affecting infants and children under the age of five, is one of the important public health issues in the Asia-Pacific region, while enteroviruses (EVs) are the etiological agents. To molecularly characterize EVs, a prospective HFMD virological surveillance program has been performed in China. In this issue, Yonghong Zhou et al. investigated the infection cases in southern China and found that the sub-genogroups CVA16_B1b, CVA6_D3a and EV-A71_C4a were the predominant enterovirus strains in 2013-2016. In addition, phylogenetic analyses of the VP1 sequences showed a long-term, population-level, persistent circulation in specific sub-genogroups in China. The cover artwork is modified from a phylogenetic tree based on VP1 sequence of EV-A71 (kindly provided by Yonghong Zhou and Hongjie Yu). See 61-74 pages for details.
|
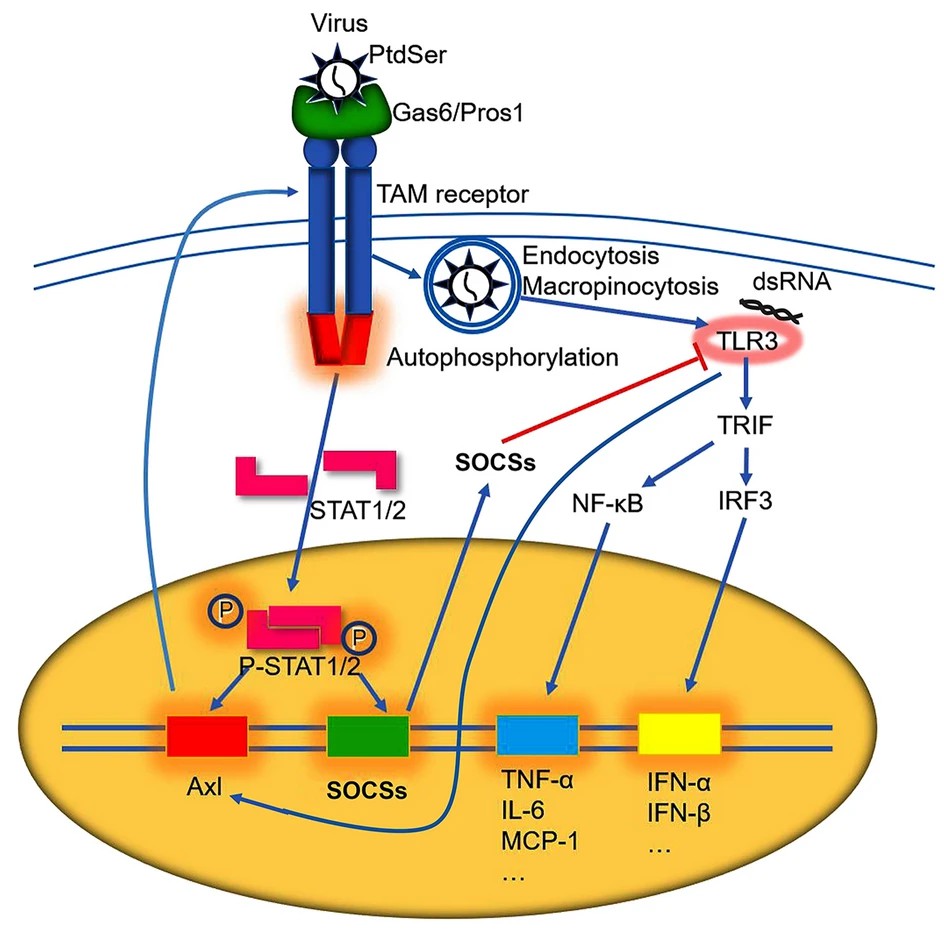
The Multifaceted Roles of TAM Receptors during Viral Infection
2021, 36(1): 1 doi: 10.1007/s12250-020-00264-9

Tyro3, Axl, and Mertk (TAM) receptors play multiple roles in a myriad of physiological and pathological processes, varying from promoting the phagocytic clearance of apoptotic cells, sustaining the immune and inflammatory homeostasis, maintaining the blood-brain barrier (BBB) integrity and central nervous system (CNS) homeostasis, to mediating cancer malignancy and chemoresistance. Growth arrest-specific protein 6 (Gas6) and protein S (Pros1) are the two ligands that activate TAM receptors. Recently, TAM receptors have been reported to mediate cell entry and infection of multitudinous enveloped viruses in a manner called apoptotic mimicry. Moreover, TAM receptors are revitalized during viral entry and infection, which sequesters innate immune and inflammatory responses, facilitating viral replication and immune evasion. However, accumulating evidence have now proposed that TAM receptors are not required for the infection of these viruses in vivo. In addition, TAM receptors protect mice against the CNS infection of neuroinvasive viruses and relieve the brain lesions during encephalitis. These protective effects are achieved through maintaining BBB integrity, attenuating pro-inflammatory cytokine production, and promoting neural cell survival. TAM receptors also regulate the programmed cell death modes of virus-infected cells, which have profound impacts on the pathogenesis and outcome of infection. Here, we systematically review the functionalities and underlying mechanisms of TAM receptors and propose the potential application of TAM agonists to prevent severe viral encephalitis.
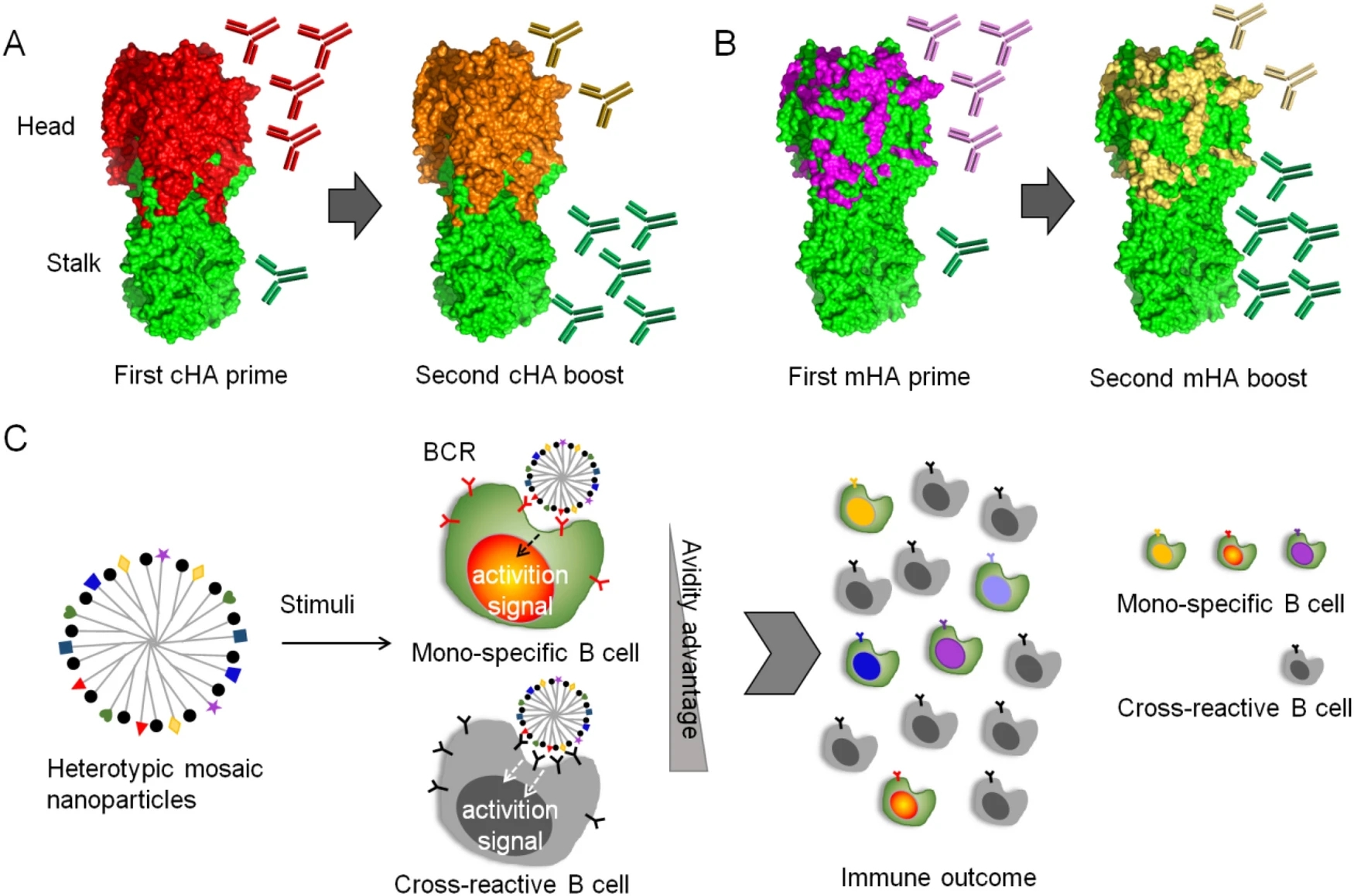
Flu Universal Vaccines: New Tricks on an Old Virus
2021, 36(1): 13 doi: 10.1007/s12250-020-00283-6
Conventional influenza vaccines are based on predicting the circulating viruses year by year, conferring limited effectiveness since the antigenicity of vaccine strains does not always match the circulating viruses. This necessitates development of universal influenza vaccines that provide broader and lasting protection against pan-influenza viruses. The discovery of the highly conserved immunogens (epitopes) of influenza viruses provides attractive targets for universal vaccine design. Here we review the current understanding with broadly protective immunogens (epitopes) and discuss several important considerations to achieve the goal of universal influenza vaccines.
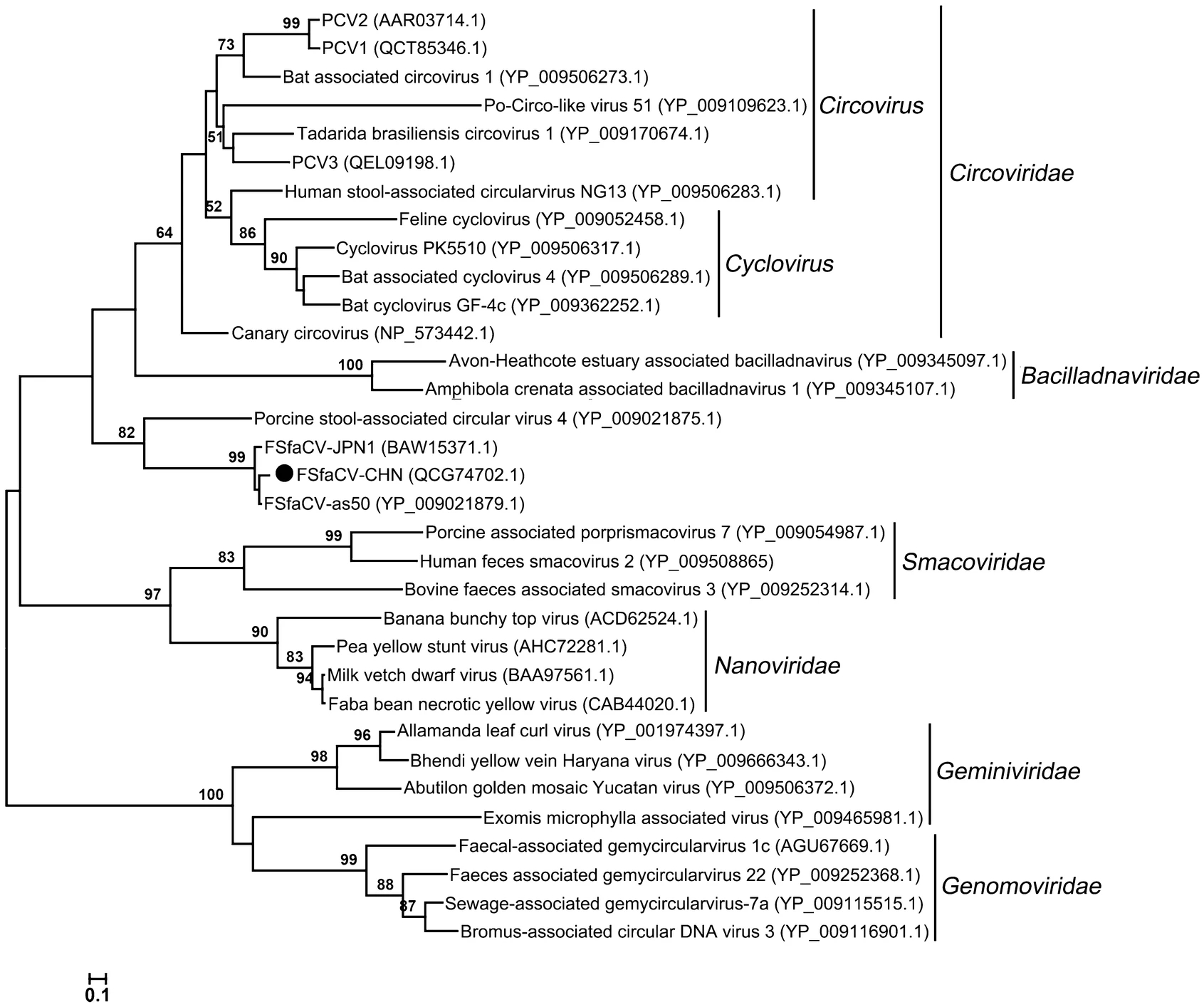
Fur Seal Feces-Associated Circular DNA Virus Identified in Pigs in Anhui, China
2021, 36(1): 25 doi: 10.1007/s12250-020-00232-3
Fur seal feces-associated circular DNA virus (FSfaCV) is an unclassified circular replication-associated protein (Rep)-encoding single-stranded (CRESS) DNA virus that has been detected in mammals (fur seals and pigs). The biology and epidemiology of the virus remain largely unknown. To investigate the virus diversity among pigs in Anhui Province, China, we pooled 600 nasal samples in 2017 and detected viruses using viral metagenomic methods. From the assembled contigs, 12 showed notably high nucleotide acid sequence similarities to the genome sequences of FSfaCVs. Based on these sequences, a full-length genome sequence of the virus was then obtained using overlapping PCR and sequencing, and the virus was designated as FSfaCV-CHN (GenBank No. MK462122). This virus shared 91.3% and 90.9% genome-wide nucleotide sequence similarities with the New Zealand fur seal strain FSfaCV-as50 and the Japanese pig strain FSfaCV-JPN1, respectively. It also clustered with the two previously identified FSfaCVs in a unique branch in the phylogenetic tree based on the open reading frame 2 (ORF2), Rep-coding gene, and the genome of the reference CRESS DNA viruses. Further epidemiological investigation using samples collected in 2018 showed that the overall positive rate for the virus was 56.4% (111/197) in Anhui Province. This is the first report of FSfaCVs identified in pigs in China, and further epidemiological studies are warranted to evaluate the influence of the virus on pigs.

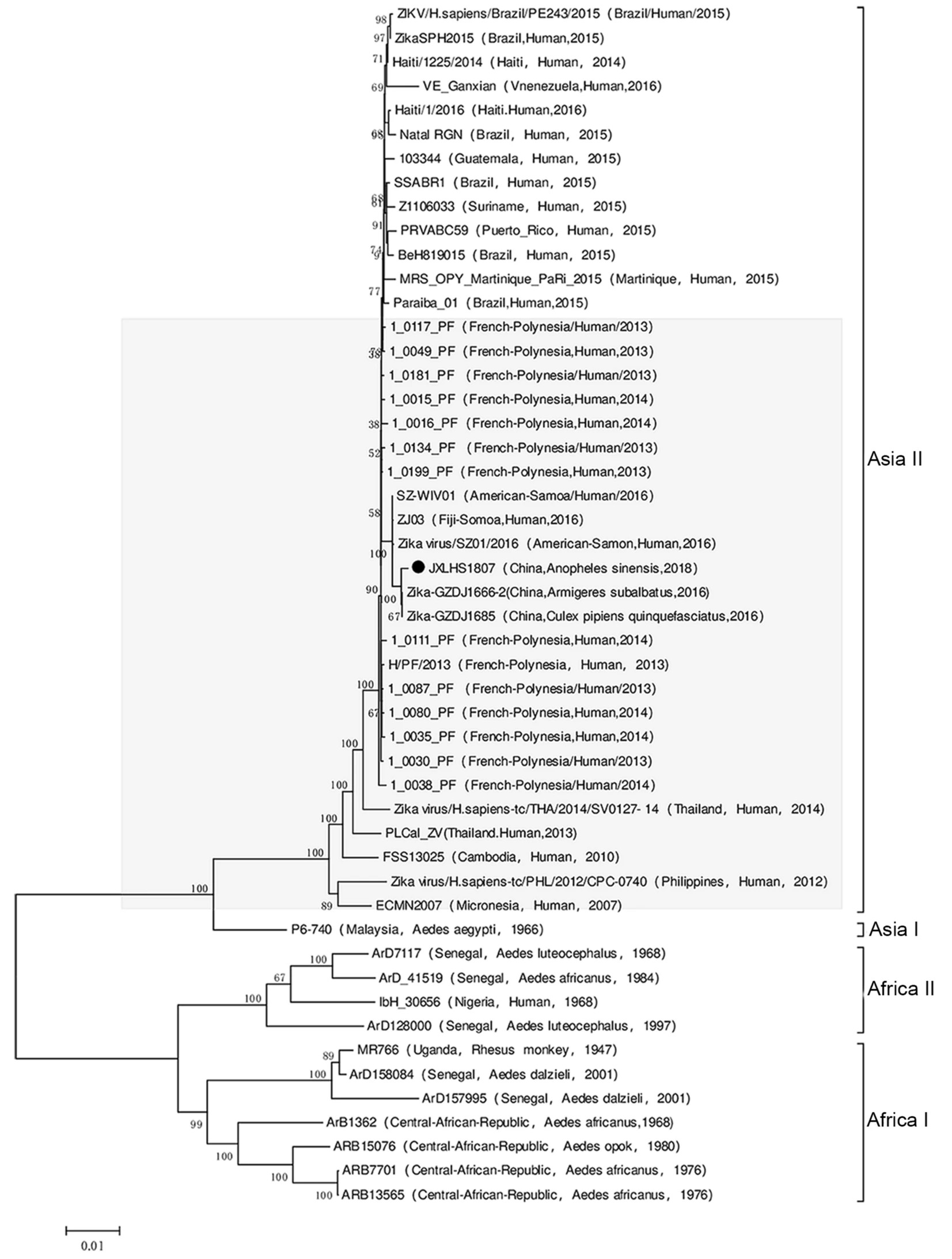
Emergence of Zika Virus in Culex tritaeniorhynchus and Anopheles sinensis Mosquitoes in China
2021, 36(1): 33 doi: 10.1007/s12250-020-00239-w
Zika virus (ZIKV) has been isolated from mosquitoes such as Aedes, Mansonia uniformis, and Culex perfuscus; However, the isolation of ZIKV from Anopheles sinensis and Culex tritaeniorhynchus has not yet been reported. In June and July 2018, 22, 985 mosquitoes and 57, 500 midges were collected in Jiangxi Province in southeastern China. Among them, six strains of ZIKV were isolated from mosquitoes: four from An. sinensis and two from Cx. tritaeniorhynchus. Molecular genetic analysis showed that the ZIKV isolated from An. sinensis and Cx. tritaeniorhynchus belonged to genotype 2 in the Asian evolutionary branch of ZIKV. In addition, the ZIKV strains isolated from An. sinensis and Cx. tritaeniorhynchus had amino acid substitutions identical to ZIKV strains prevalent in South America since 2015. This study is the first to isolate ZIKV from mosquito specimens collected in the wild of Jiangxi Province, China; This is also the first time that ZIKV has been isolated from An. sinensis and Cx. tritaeniorhynchus. Given that An. sinensis and Cx. tritaeniorhynchus have a very wide geographical distribution in China and even in eastern and southern Asia, the isolation of several strains of ZIKV from these two mosquitoes poses new challenges for the prevention and control of ZIKV infection in the mainland of China and countries and regions with the same distribution of mosquitoes.
Epidemiology and Genotypic Diversity of Eurasian Avian-Like H1N1 Swine Influenza Viruses in China
2021, 36(1): 43 doi: 10.1007/s12250-020-00257-8
Eurasian avian-like H1N1 (EA H1N1) swine influenza virus (SIV) outside European countries was first detected in Hong Kong Special Administrative Region (Hong Kong, SAR) of China in 2001. Afterwards, EA H1N1 SIVs have become predominant in pig population in this country. However, the epidemiology and genotypic diversity of EA H1N1 SIVs in China are still unknown. Here, we collected the EA H1N1 SIVs sequences from China between 2001 and 2018 and analyzed the epidemic and phylogenic features, and key molecular markers of these EA H1N1 SIVs. Our results showed that EA H1N1 SIVs distributed in nineteen provinces/municipalities of China. After a long-time evolution and transmission, EA H1N1 SIVs were continuously reassorted with other co-circulated influenza viruses, including 2009 pandemic H1N1 (A(H1N1)pdm09), and triple reassortment H1N2 (TR H1N2) influenza viruses, generated 11 genotypes. Genotype 3 and 5, both of which were the reassortments among EA H1N1, A(H1N1)pdm09 and TR H1N2 viruses with different origins of M genes, have become predominant in pig population. Furthermore, key molecular signatures were identified in EA H1N1 SIVs. Our study has drawn a genotypic diversity image of EA H1N1 viruses, and could help to evaluate the potential risk of EA H1N1 for pandemic preparedness and response.
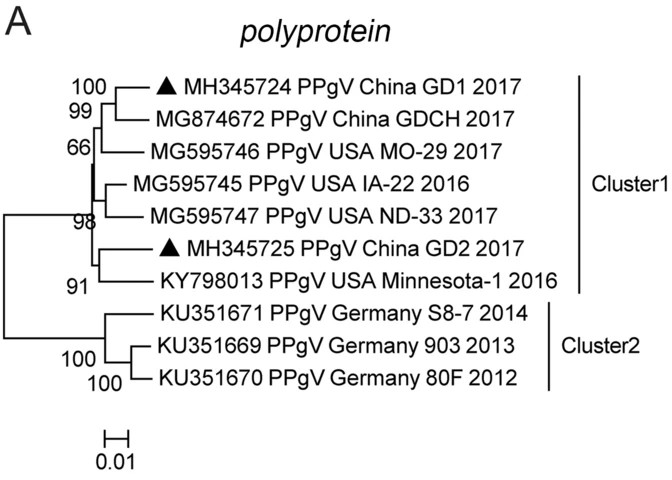
The Prevalence, Genetic Characterization, and Evolutionary Analysis of Porcine Pegivirus in Guangdong, China
2021, 36(1): 52 doi: 10.1007/s12250-020-00240-3
Porcine pegivirus (PPgV) is a member of the Pegivirus genus in the Flaviviridae family. PPgV is an emerging virus that has been discovered in swine herds in Germany, the United States, China, Poland, Italy, and the United Kingdom, indicating a wide geographical distribution. In this retrospective study, 339 pig serum samples were collected from 20 different commercial swine farms located in nine cities in Guangdong Province, China, from 2016 to 2018, to investigate the prevalence and genetic diversity of PPgV in this geographical region. PPgV was detected in 55% (11/20) of the farms using nested reverse transcription PCR, with 6.2% (21/339) of pigs testing positive for PPgV. The yearly PPgV-positive rate increased from 2.6% to 7.5% between 2016 and 2018. Sequencing of PPgV-positive samples identified two complete polyprotein genes and seven partial NS5B genes from different farms. Comparative analysis of the polyprotein genes revealed that PPgV sequences obtained in this study showed 87.4%–97.2% similarity at the nucleotide level and 96.5%–99.4% similarity at the amino acid level with the reference sequences. Sequence alignment and phylogenetic analysis of the complete polyprotein gene and partial NS5B and NS3 genes demonstrated a high genetic similarity with the samples from the USA. The finding of the wide distribution of PPgV in swine herds in Guangdong Province will contribute to the understanding of the epidemiological characteristics and genetic evolution of PPgV in China.
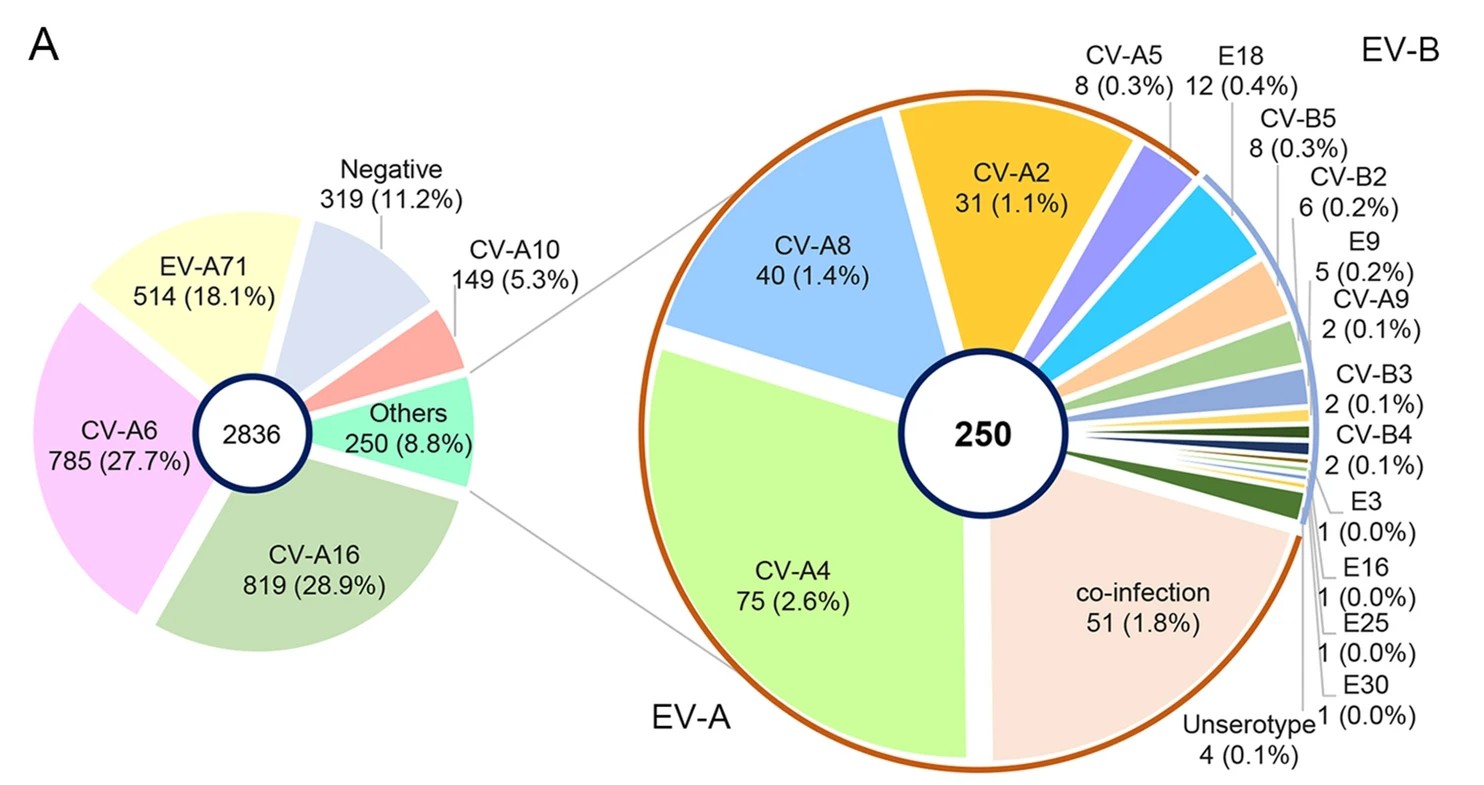
Genetic Variation of Multiple Serotypes of Enteroviruses Associated with Hand, Foot and Mouth Disease in Southern China
2021, 36(1): 61 doi: 10.1007/s12250-020-00266-7
Enteroviruses (EVs) species A are a major public health issue in the Asia–Pacific region and cause frequent epidemics of hand, foot and mouth disease (HFMD) in China. Mild infections are common in children; however, HFMD can also cause severe illness that affects the central nervous system. To molecularly characterize EVs, a prospective HFMD virological surveillance program was performed in China between 2013 and 2016. Throat swabs, rectal swabs and stool samples were collected from suspected HFMD patients at participating hospitals. EVs were detected using generic real-time and nested reverse transcription-polymerase chain reactions (RT-PCRs). Then, the complete VP1 regions of enterovirus A71 (EV-A71), coxsackievirus A16 (CVA16) and CVA6 were sequenced to analyze amino acid changes and construct a viral molecular phylogeny. Of the 2836 enrolled HFMD patients, 2, 517 (89%) were EV positive. The most frequently detected EVs were CVA16 (32.5%, 819), CVA6 (31.2%, 785), and EV-A71 (20.4%, 514). The subgenogroups CVA16_B1b, CVA6_D3a and EV-A71_C4a were predominant in China and recombination was not observed in the VP1 region. Sequence analysis revealed amino acid variations at the 30, 29 and 44 positions in the VP1 region of EV-A71, CVA16 and CVA6 (compared to the respective prototype strains BrCr, G10 and Gdula), respectively. Furthermore, in 21 of 24 (87.5%) identified EV-A71 samples, a known amino acid substitution (D31N) that may enhance neurovirulence was detected. Our study provides insights about the genetic characteristics of common HFMD-associated EVs. However, the emergence and virulence of the described mutations require further investigation.
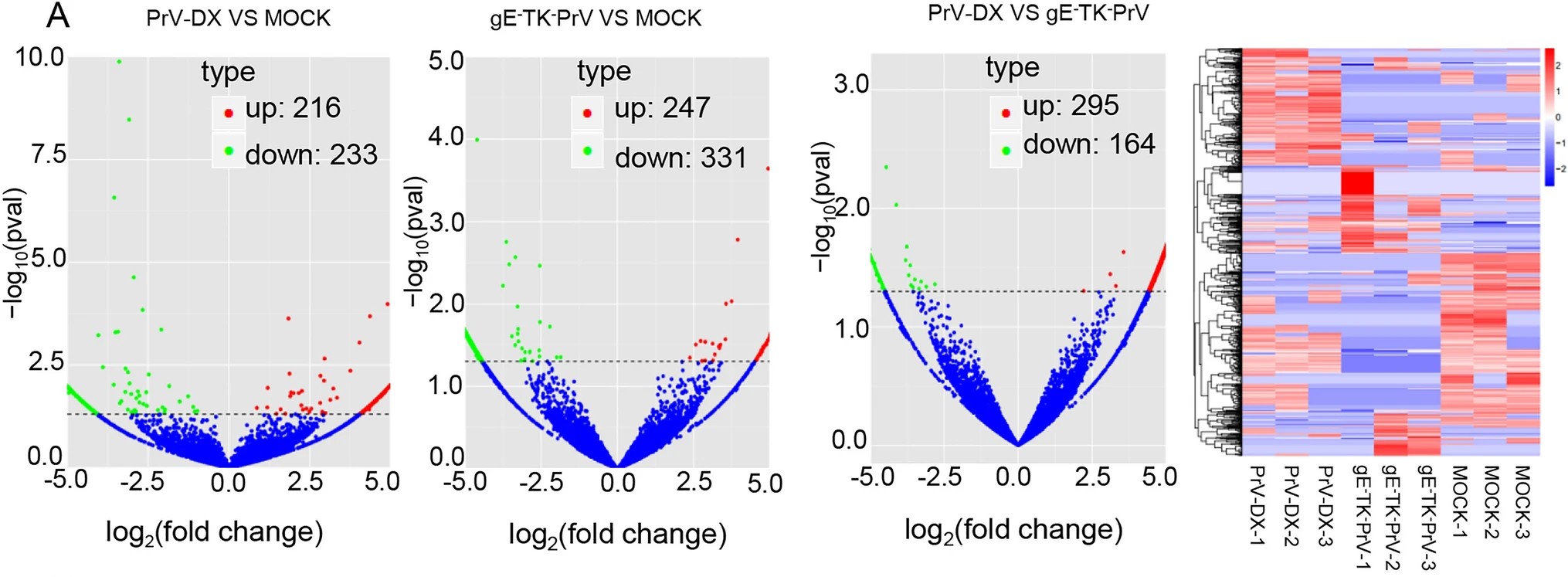
Differential CircRNA Expression Profiles in PK-15 Cells Infected with Pseudorabies Virus Type Ⅱ
2021, 36(1): 75 doi: 10.1007/s12250-020-00255-w
Circular RNAs (circRNAs) belong to a class of non-coding RNAs with diverse biological functions. However, little is known about their roles in case of pseudorabies virus (PrV) infection. Here, we analyzed the expression profile of host circRNAs from a virulent PrV type Ⅱ strain DX (PrV-DX) infected and an attenuated gE/TK deficient (gE-TK-PrV) strain of PrV infected PK-15 cells. CircRNAs were identified by find_circ and analyzed with DESeq 2. Compared with the mock cells, 449 differentially expressed (DE) circRNAs (233 down-regulated and 216 up-regulated) from PrV-DX infected and 578 DE circRNAs (331 down-regulated and 247 up-regulated) from gE-TK-PrV infected PK-15 cells were identified. In addition, 459 DE circRNAs (164 down-regulated and 295 up-regulated) between the PrV-DX and gE-TK-PrV infected cells were identified. The expression patterns of 13 circRNAs were validated by reverse transcription quantitative real-time PCR (RT-qPCR) and results were similar as of RNA-seq. The putative target miRNA binding sites of DE circRNAs were predicted by using miRanda and psRobot. The circRNA-miRNA-mRNA network was constructed and certain miRNAs that have possible roles in antiviral immune response, such as miR-210 and miR-340, were predicted. GO and KEGG pathway analysis demonstrated that DE circRNAs were enriched in the processes such as cellular metabolism, protein binding, RNA degradation and regulation of actin cytoskeleton. Collectively, these findings might provide the useful information for a better understanding of mechanisms underlying the interaction between PrV-Ⅱ and host cells.
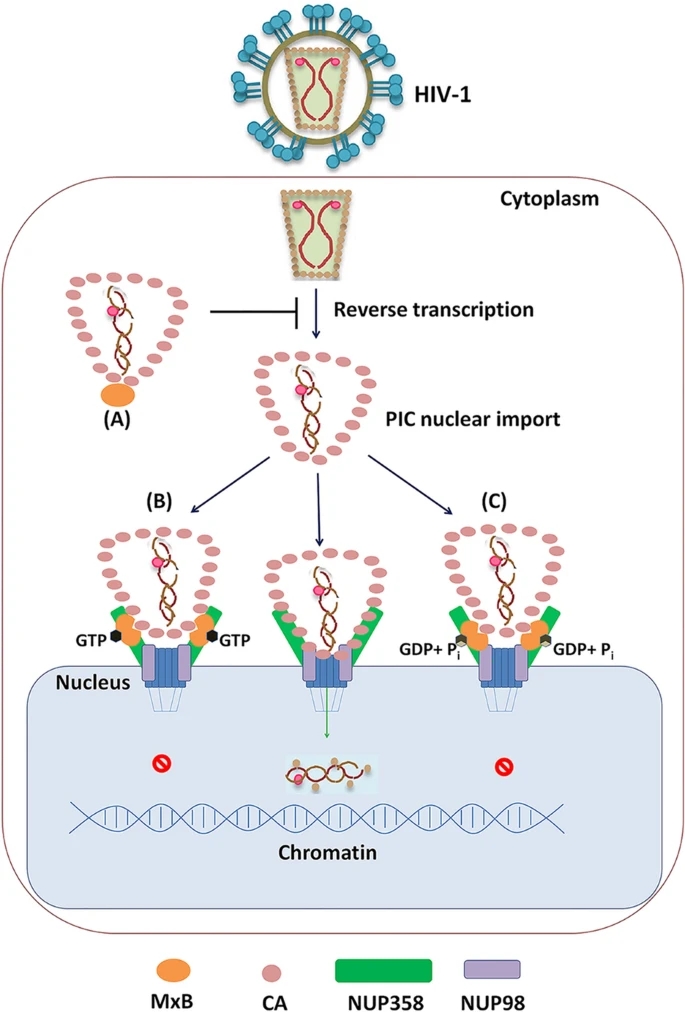
GTPase Activity of MxB Contributes to Its Nuclear Location, Interaction with Nucleoporins and Anti-HIV-1 Activity
2021, 36(1): 85 doi: 10.1007/s12250-020-00249-8
The human myxovirus resistance 2 (Mx2/MxB) protein, a member of interferon (IFN)-inducible dynamin-like large GTPases, restricts a number of virus infections. Inhibition of these viruses occurs at poorly-defined steps after viral entry and has a common requirement for MxB oligomerization. However, the GTPase activity is essential for the anti-viral effects of MxB against herpesviruses and HBV but not HIV-1. To understand the role of MxB GTPase activity, including GTP binding and GTP hydrolysis, in restriction of HIV-1 infection, we genetically separated these two functions and evaluated their contributions to restriction. We found that both the GTP binding and hydrolysis function of MxB involved in the restriction of HIV-1 replication. The GTPase activity of MxB contributed to its nuclear location, interaction with nucleoporins (NUPs) and HIV-1 capsids. Furthermore, MxB disrupted the association between NUPs and HIV-1 cores dependently upon its GTPase activity. The function of GTPase activity was therefore multi-faceted, led to fundamentally distinct mechanisms employed by wild-type MxB and GTPase activity defective MxB mutations to restrict HIV-1 replication.
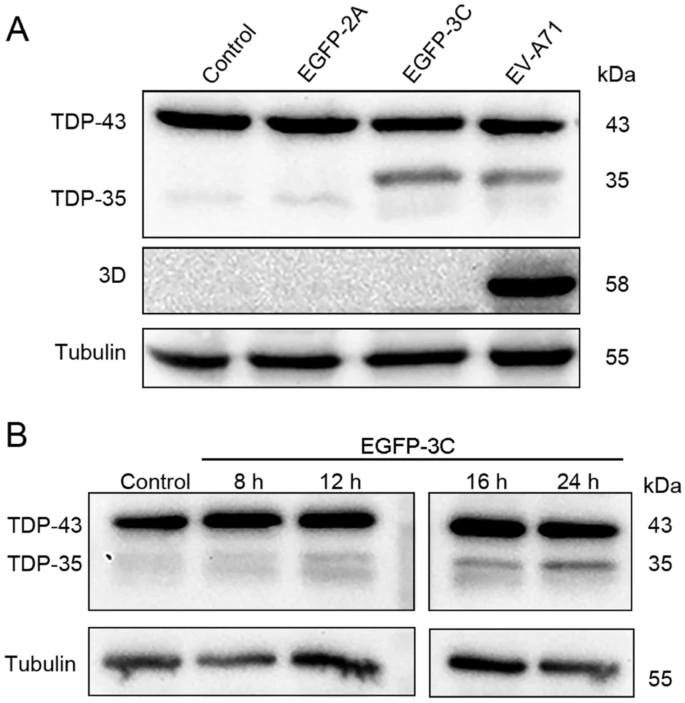
TAR DNA-Binding Protein 43 is Cleaved by the Protease 3C of Enterovirus A71
2021, 36(1): 95 doi: 10.1007/s12250-020-00262-x
Enterovirus A71 (EV-A71) is one of the etiological pathogens leading to hand, foot, and mouth disease (HFMD), which can cause severe neurological complications. The neuropathogenesis of EV-A71 infection is not well understood. The mislocalization and aggregation of TAR DNA-binding protein 43 (TDP-43) is the pathological hallmark of amyotrophic lateral sclerosis (ALS). However, whether TDP-43 was impacted by EV-A71 infection is unknown. This study demonstrated that TDP-43 was cleaved during EV-A71 infection. The cleavage of TDP-43 requires EV-A71 replication rather than the activated caspases due to viral infection. TDP-43 is cleaved by viral protease 3C between the residues 331Q and 332S, while mutated TDP-43 (Q331A) was not cleaved. In addition, mutated 3C which lacks the protease activity failed to induce TDP-43 cleavage. We also found that TDP-43 was translocated from the nucleus to the cytoplasm, and the mislocalization of TDP-43 was induced by viral protease 2A rather than 3C. Taken together, we demonstrated that TDP-43 was cleaved by viral protease and translocated to the cytoplasm during EV-A71 infection, implicating the possible involvement of TDP-43 in the pathogenesis of EV-A71infection.
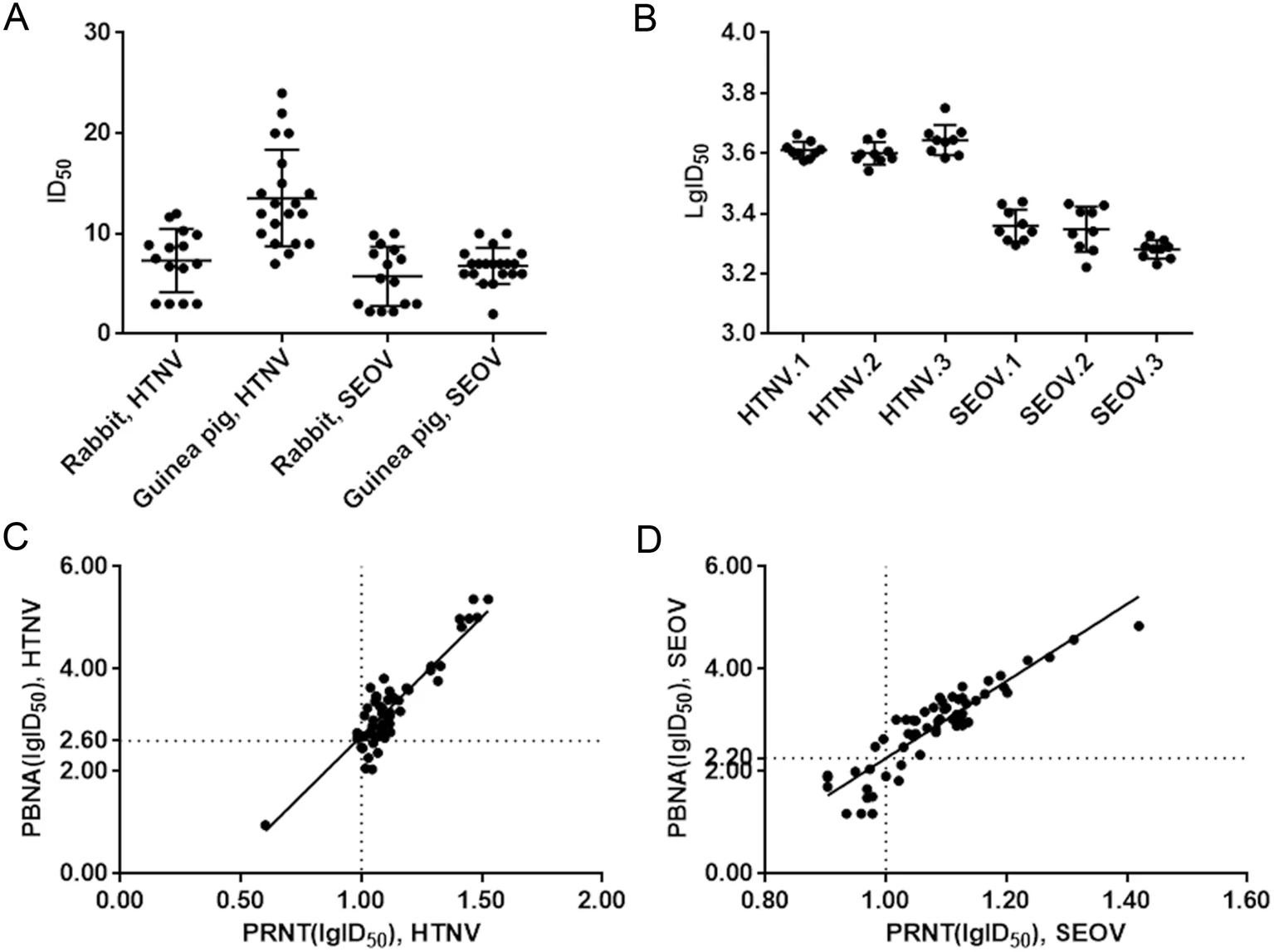
Monitoring Neutralization Property Change of Evolving Hantaan and Seoul Viruses with a Novel Pseudovirus-Based Assay
2021, 36(1): 104 doi: 10.1007/s12250-020-00237-y
The Hantaan virus (HTNV) and Seoul virus (SEOV) mutants have accumulated over time. It is important to determine whether their neutralizing epitopes have evolved, thereby making the current vaccine powerless. However, it is impossible to determine by using traditional plaque reduction neutralization test (PRNT), because it requires large numbers of live mutant strains. Pseudovirus-based neutralization assays (PBNA) were developed by employing vesicular stomatitis virus (VSV) backbone incorporated with HTNV or SEOV glycoproteins (VSVΔG*-HTNVG or VSVΔG*-SEOVG). 56 and 51 single amino acid substitutions of glycoprotein (GP) in HTNV and SEOV were selected and introduced into the reference plasmid. Then the mutant pseudoviruses were generated and tested by PBNA. The PBNA results were highly correlated with PRNT ones with R2 being 0.91 for VSVΔG*-HTNVG and 0.82 for VSVΔG*-SEOVG. 53 HTNV mutant pseudoviruses and 46 SEOV mutants were successfully generated. Importantly, by using PBNA, we found that HTNV or SEOV immunized antisera could neutralize all the corresponding 53 HTNV mutants or the 46 SEOV mutants respectively. The novel PBNA enables us to closely monitor the effectiveness of vaccines against large numbers of evolving HTNV and SEOV. And the current vaccine remains to be effective for the naturally occurring mutants.
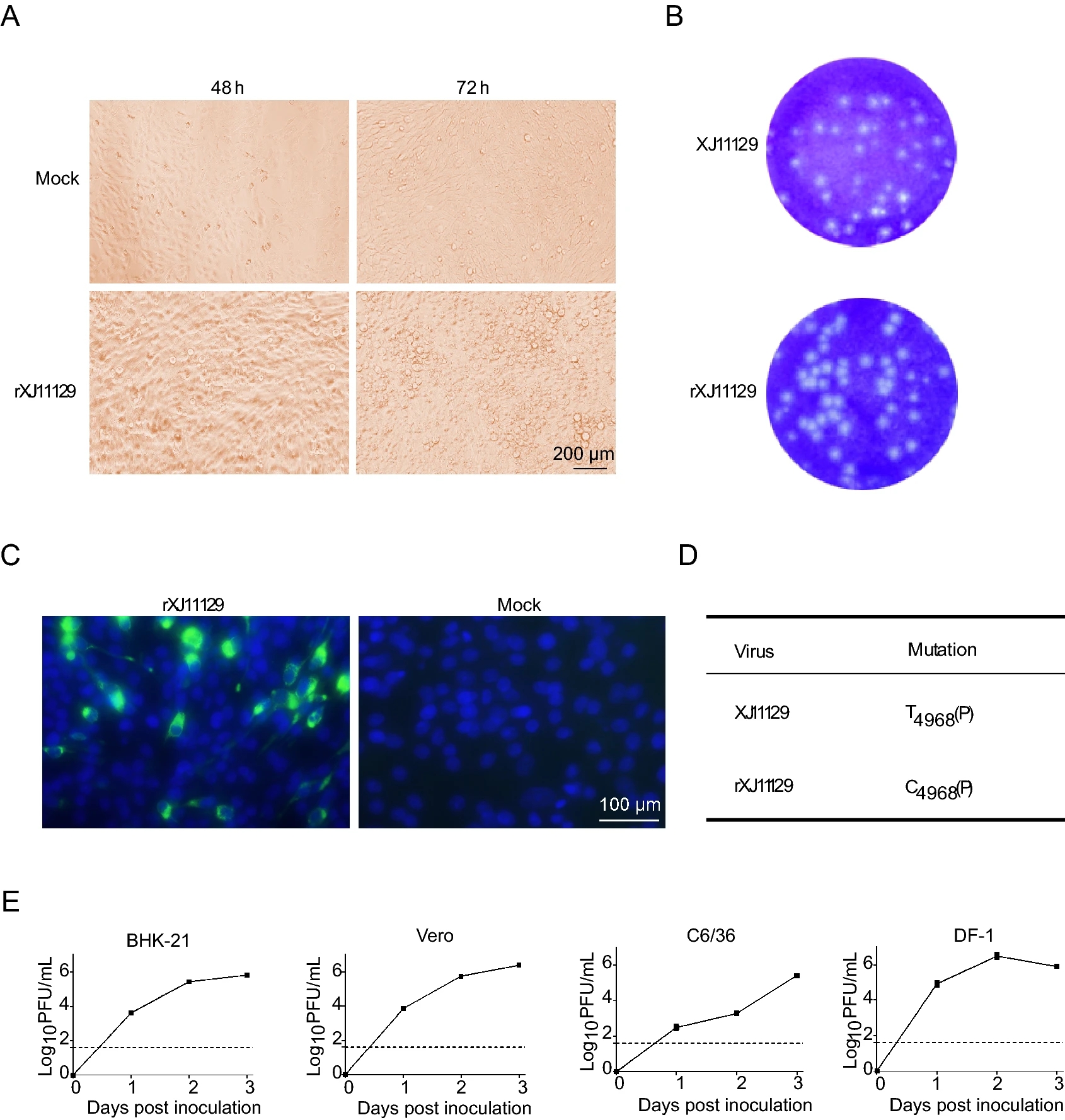
Recovery and Genetic Characterization of a West Nile Virus Isolate from China
2021, 36(1): 113 doi: 10.1007/s12250-020-00246-x
West Nile virus (WNV) is an important neurotropic flavivirus that is widely distributed globally. WNV strain XJ11129 was first isolated in Xinjiang, China, and its genetic and biological characteristics remain largely unknown. In this study, phylogenetic and sequence analyses revealed that XJ11129 belongs to lineage 1a and shares high genetic identity with the highly pathogenic strain NY99. Then, the full-length genomic cDNA of XJ11129 was amplified and assembled using a modified Gibson assembly (GA) method. The virus (named rXJ11129) was successfully rescued in days following this method. Compared with other wild-type WNV isolates, rXJ11129 exhibited virulence indistinguishable from that of the NY99 strain in vivo. In summary, the genomic and virulence phenotypes of rXJ11129 were characterized in vivo and in vitro, and these data will improve the understanding of the spread and pathogenesis of this reemerging virus.
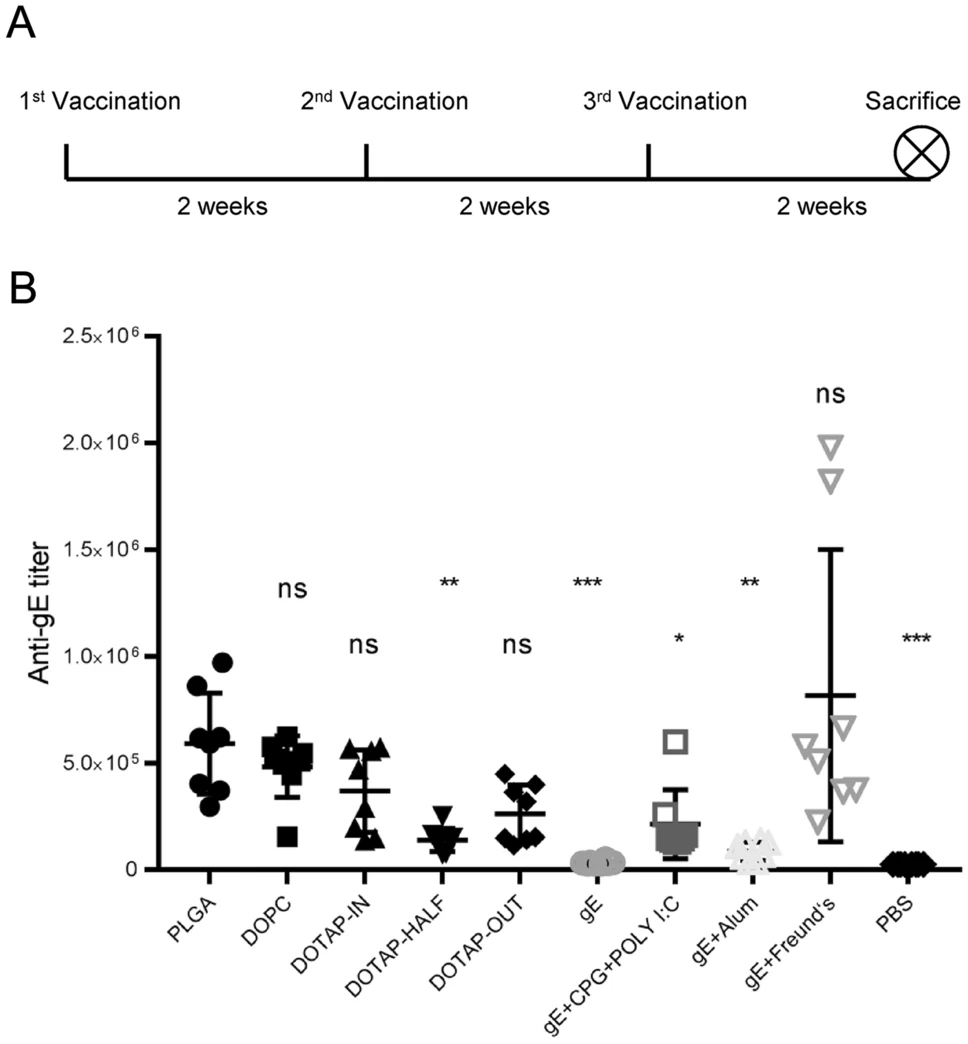
Immune Responses to Varicella-Zoster Virus Glycoprotein E Formulated with Poly(Lactic-co-Glycolic Acid) Nanoparticles and Nucleic Acid Adjuvants in Mice
2021, 36(1): 122 doi: 10.1007/s12250-020-00261-y
The subunit herpes zoster vaccine Shingrix is superior to attenuated vaccine Zostavax in both safety and efficacy, yet its unlyophilizable liposome delivery system and the limited supply of naturally sourced immunological adjuvant QS-21 still need to be improved. Based on poly(lactic-co-glycolic acid) (PLGA) delivery systems that are stable during the lyophilization and rehydration process and using a double-emulsion (w/o/w) solvent evaporation method, we designed a series of nanoparticles with varicella-zoster virus antigen glycoprotein E (VZV-gE) as an antigen and nucleic acids including polyinosinic-polycytidylic acid (Poly I:C) and phosphodiester CpG oligodeoxynucleotide (CpG ODN), encapsulated as immune stimulators. While cationic lipids (DOTAP) have more potential than neutral lipids (DOPC) for activating gE-specific cell-mediated immunity (CMI) in immunized mice, especially when gE is encapsulated in and presented on the surface of nanoparticles, PLGA particles without lipids have the greatest potential to induce not only the highest gE-specific IgG titers but also the strongest gE-specific CMI responses, including the highest proportions of interferon-γ (IFN-γ)- and interleukin-2 (IL-2)-producing CD4+/CD8+T cells according to a flow cytometry assay and the greatest numbers of IFN-γ- and IL-2-producing splenocytes according to an enzyme-linked immunospot (ELISPOT) assay. These results showed that immune-stimulating nucleic acids together with the PLGA delivery system showed promise as a safe and economical varicella and zoster vaccine candidate.
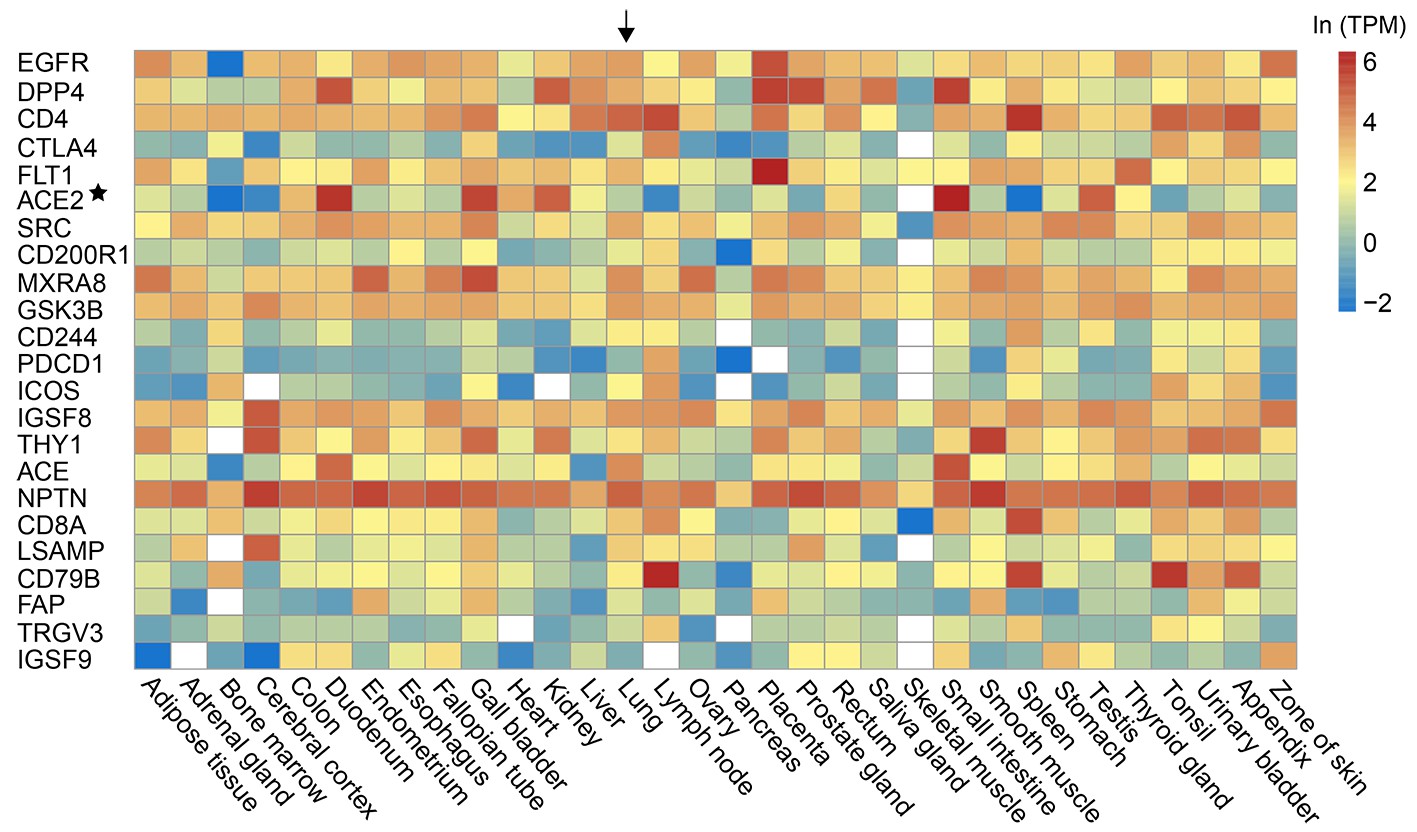
Prediction of the Receptorome for the Human-Infecting Virome
2021, 36(1): 133 doi: 10.1007/s12250-020-00259-6
The virus receptors are key for the viral infection of host cells. Identification of the virus receptors is still challenging at present. Our previous study has shown that human virus receptor proteins have some unique features including high N-glycosylation level, high number of interaction partners and high expression level. Here, a random-forest model was built to identify human virus receptorome from human cell membrane proteins with an accepted accuracy based on the combination of the unique features of human virus receptors and protein sequences. A total of 1424 human cell membrane proteins were predicted to constitute the receptorome of the human-infecting virome. In addition, the combination of the random-forest model with protein-protein interactions between human and viruses predicted in previous studies enabled further prediction of the receptors for 693 human-infecting viruses, such as the enterovirus, norovirus and West Nile virus. Finally, the candidate alternative receptors of the SARS-CoV-2 were also predicted in this study. As far as we know, this study is the first attempt to predict the receptorome for the human-infecting virome and would greatly facilitate the identification of the receptors for viruses.
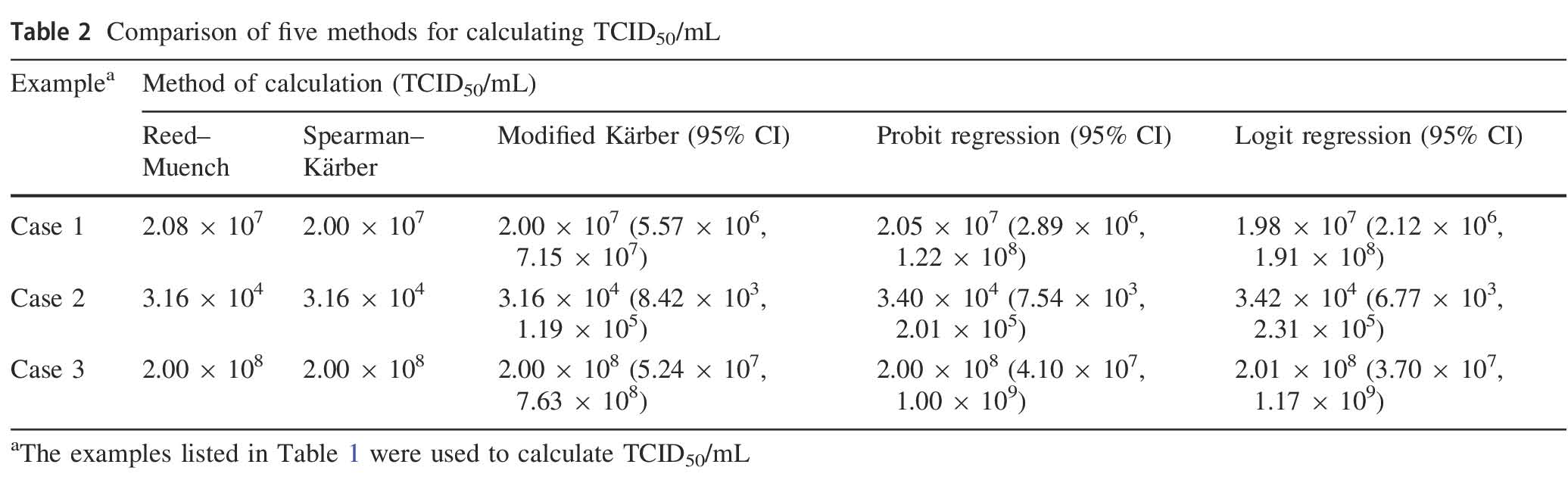
On the Calculation of TCID50 for Quantitation of Virus Infectivity
2021, 36(1): 141 doi: 10.1007/s12250-020-00230-5
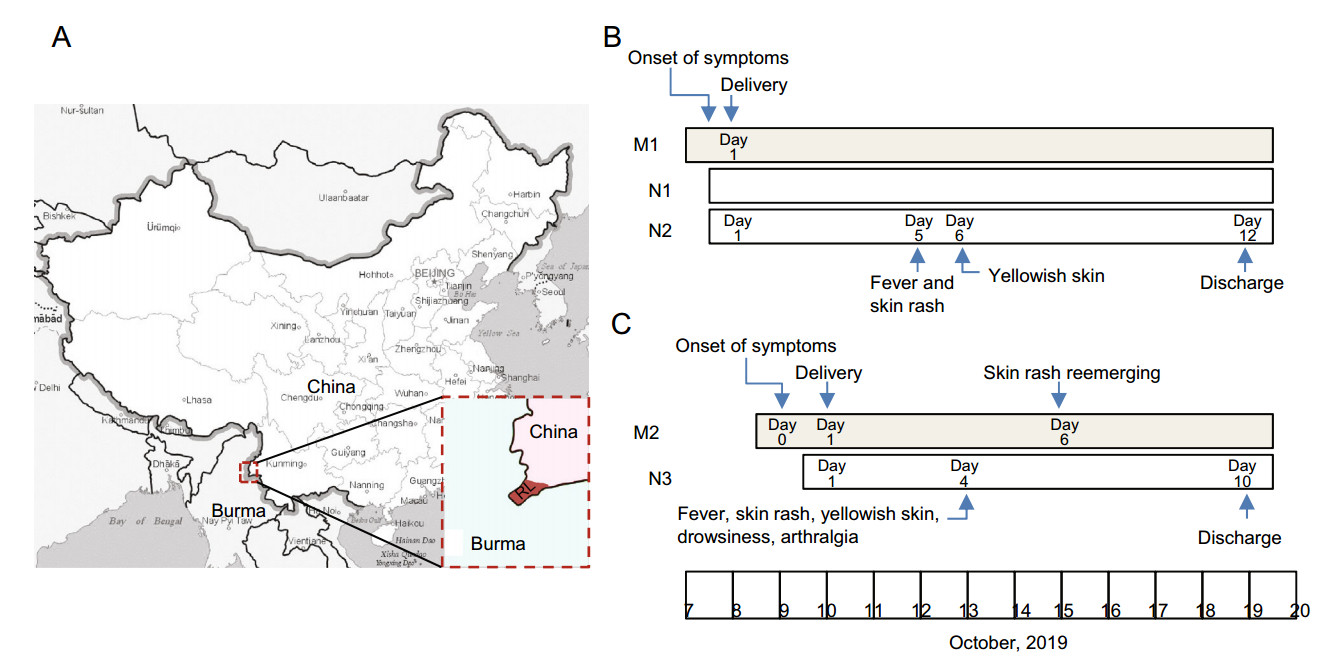
Perinatal Vertical Transmission of Chikungunya Virus in Ruili, a Town on the Border between China and Myanmar
2021, 36(1): 145 doi: 10.1007/s12250-020-00245-y

Novel HCV-Like Virus Detected in Avian Livers in Southern China and Its Implications for Natural Recombination Events
2021, 36(1): 149 doi: 10.1007/s12250-020-00256-9
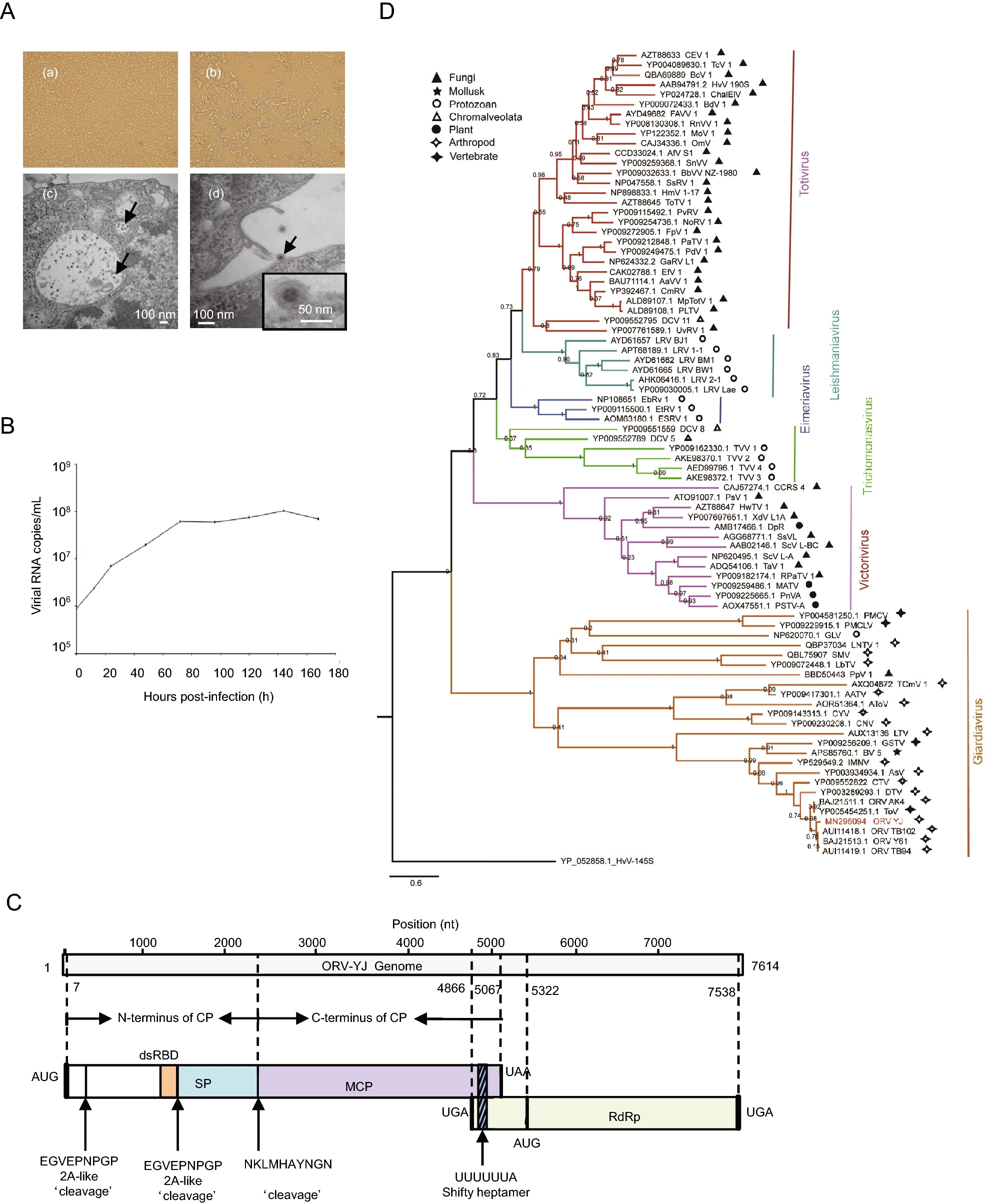
Identification and Molecular Characterization of a New Omono River Virus Isolated from Culex Tritaeniorhynchus in Yunnan, China
2021, 36(1): 152 doi: 10.1007/s12250-020-00247-w
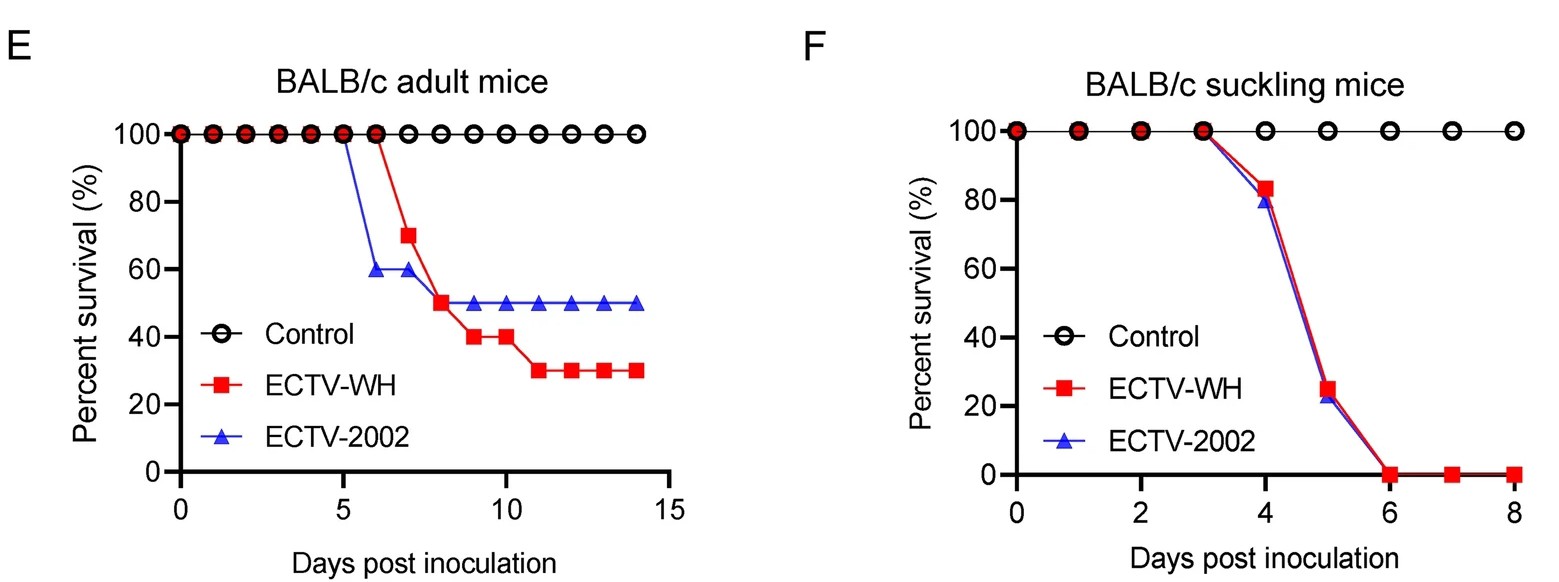
Identification, Isolation, and Characterization of an Ectromelia Virus New Strain from an Experimental Mouse
2021, 36(1): 155 doi: 10.1007/s12250-020-00263-w

Concerns on Vaccine against Varicella Caused by Varicella-Zoster Virus Infection
2021, 36(1): 159 doi: 10.1007/s12250-020-00231-4
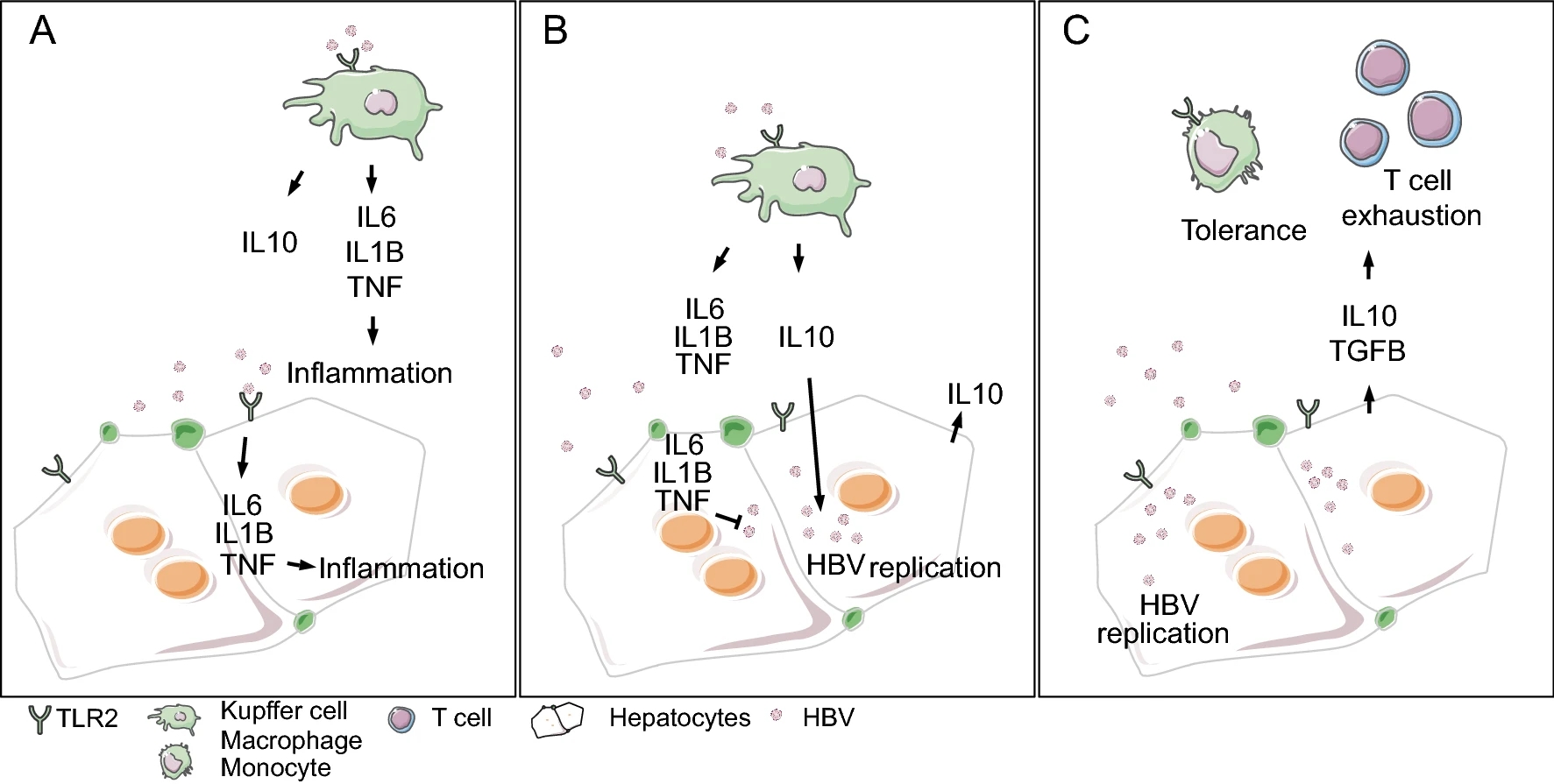
Controversial: Early Innate Responses to Hepatitis B Virus Infection, an Explanation for Viral Persistence?
2021, 36(1): 163 doi: 10.1007/s12250-020-00235-0