In recent decades, bats have been identified as natural hosts of various zoonotic viruses. Bat-borne viruses, directly or indirectly spilling into human society, have caused numerous devastating outbreaks. To prevent future pandemics, it is crucial to conduct long-term epidemiological surveys on natural reservoirs that have close interactions with humans and livestock. In this issue, Guo et al. investigated the molecular characteristics and genetic diversity of coronaviruses discovered in different bat species from southern China. Two representative full-length genomes of bat CoVs were obtained, which share the highest identity with pangolin HKU4-related coronaviruses. One of the strains has a putative furin protease cleavage site in its S protein and is likely to utilize human dipeptidyl peptidase-4 as a cell-entry receptor. This is the first report of a bat HKU4-related-CoV strain containing a furin protease cleavage site. The cover image depicts the greater bamboo bat sharing its habitat with other wild animals that live near human villages and have frequent interactions with humans, livestock, and poultry. (Kindly designed and provided by Prof. Libiao Zhang). See page 868–876 for details.
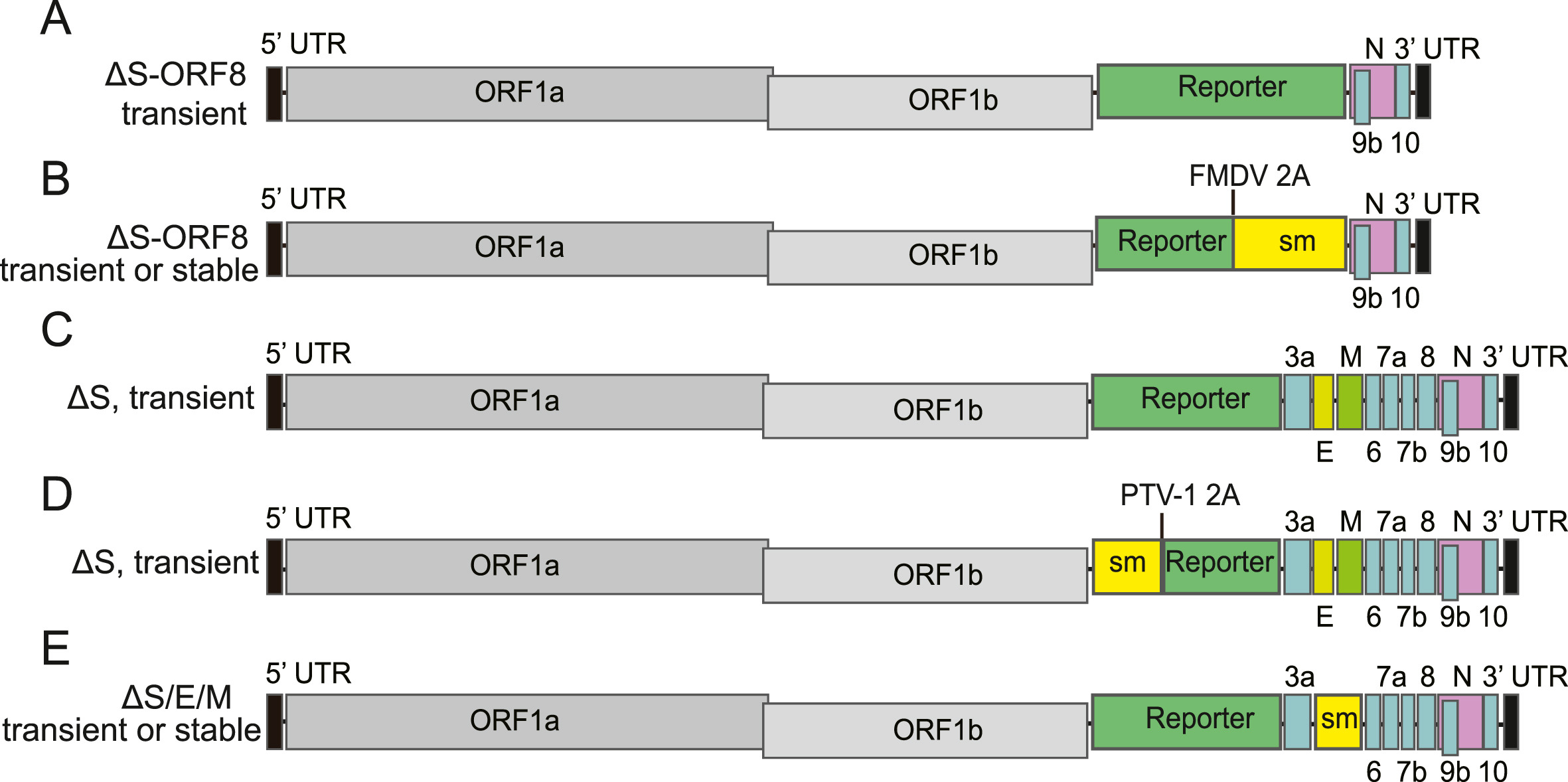
The recent emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) caused serious harm to human health and struck a blow to global economic development. Research on SARS-CoV-2 has greatly benefited from the use of reverse genetics systems, which have been established to artificially manipulate the viral genome, generating recombinant and reporter infectious viruses or biosafety level 2 (BSL-2)-adapted non-infectious replicons with desired modifications. These tools have been instrumental in studying the molecular biological characteristics of the virus, investigating antiviral therapeutics, and facilitating the development of attenuated vaccine candidates. Here, we review the construction strategies, development, and applications of reverse genetics systems for SARS-CoV-2, which may be applied to other CoVs as well.
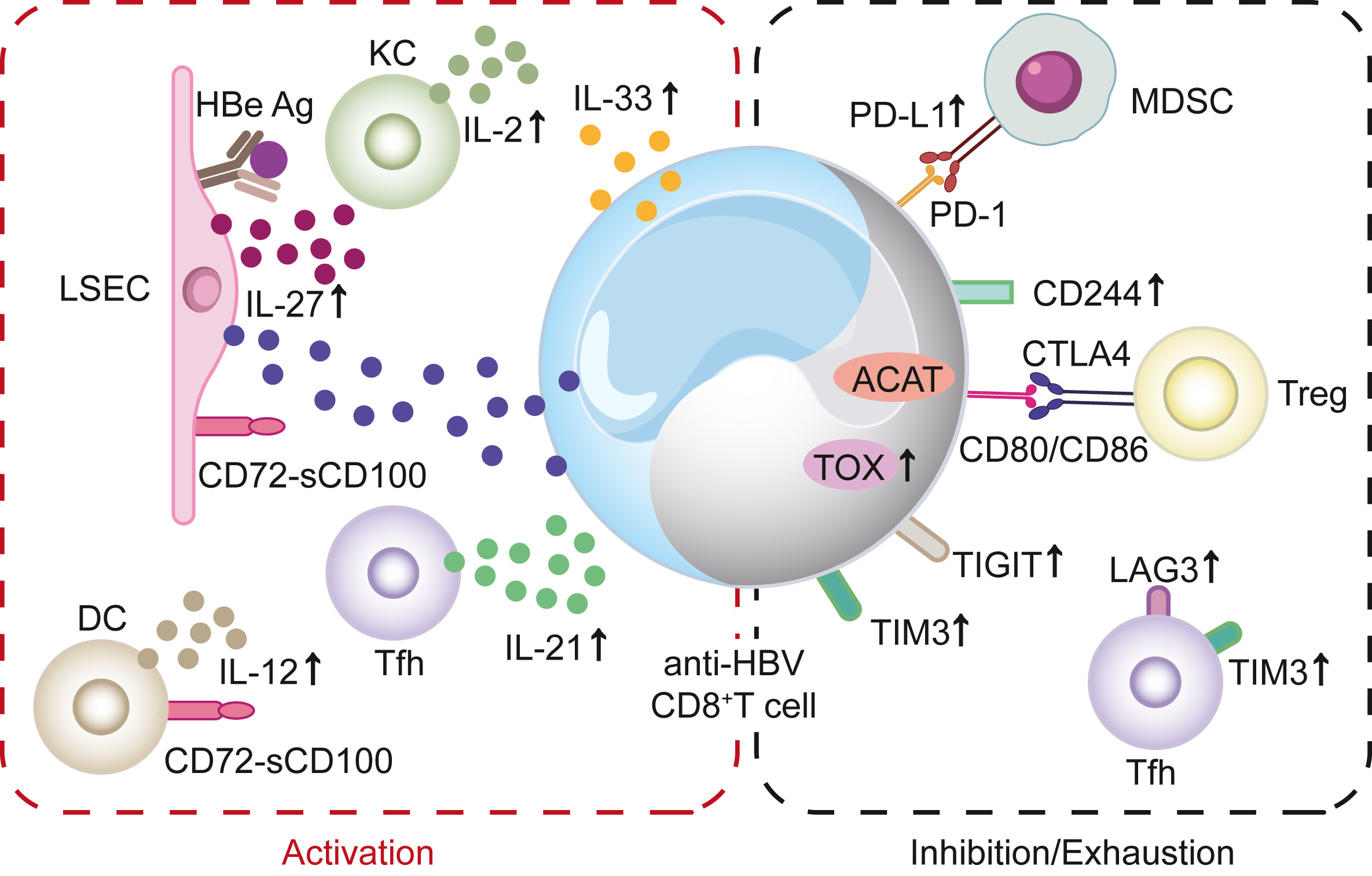
Chronic hepatitis B virus (HBV) infection remains a major public health concern globally, and T cell responses are widely believed to play a pivotal role in mediating HBV clearance. Accordingly, research on the characteristics of HBV-specific T cell responses, from activation to exhaustion, has advanced rapidly. Here, we summarize recent developments in characterizing T cell immunity in HBV infection by reviewing basic and clinical research published in the last five years. We provide a comprehensive summary of the mechanisms that induce effective anti-HBV T cell immunity, as well as the latest developments in understanding T cell dysfunction in chronic HBV infection. Furthermore, we briefly discuss current novel treatment strategies aimed at restoring anti-HBV T cell responses.
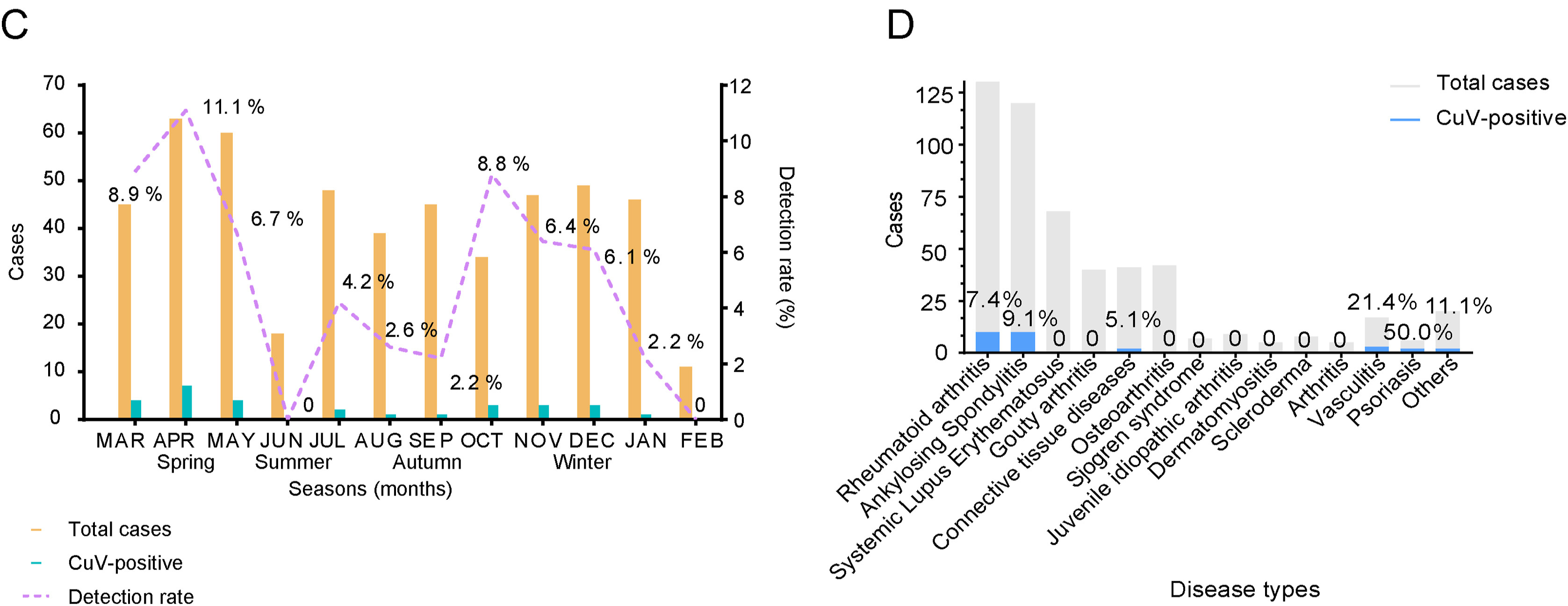
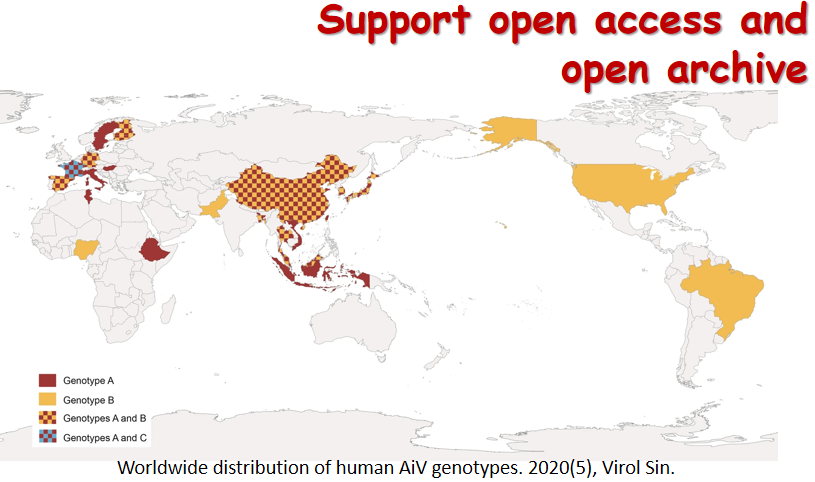
Cutavirus (CuV) is a novel protoparvovirus possibly associated with diarrhea and cutaneous T-cell lymphomas. Patients with rheumatic disease are immunosuppressed and may be more vulnerable to pathogenic viruses. A descriptive study was conducted among hospitalized patients with rheumatic diseases and individuals undergoing medical health check-ups between June 2019 and June 2022 in Guangzhou, China. Stool samples of subjects were tested for CuV DNA. Demographic and fecal examination data of patients were obtained from electronic medical records. A total of 505 patients with rheumatic diseases and 244 individuals who underwent medical health check-ups were included in the study. Of the patients with rheumatic disease, 5.74% [95% confidence interval (CI): 4.03%–8.12%] were positive for CuV DNA, while no individual in the medical health check-up group was positive, indicating a close correlation between CuV and rheumatic disease. Men and patients with rheumatoid arthritis or ankylosing spondylitis, according to the disease classification, were more susceptible to being infected with CuV (P < 0.01). After adjustments, being male remained the only significant factor, with an adjusted odd ratio (OR) of 4.4 (95% CI: 1.7–11.4, P = 0.002). Phylogenetic analysis of the CuV VP2 sequences showed three diverse clades, one of which was segregated to be a single branching independent of previously known sequences, which is possible a new genotype.
Coronavirus (CoV) spillover originating from game animals, particularly pangolins, is currently a significant concern. Meanwhile, vigilance is urgently needed for coronaviruses carried by bats, which are known as natural reservoirs of many coronaviruses. In this study, we collected 729 anal swabs of 20 different bat species from nine locations in Yunnan and Guangdong provinces, southern China, in 2016 and 2017, and described the molecular characteristics and genetic diversity of alphacoronaviruses (αCoVs) and betacoronaviruses (βCoVs) found in these bats. Using RT-PCR, we identified 58 (8.0%) bat CoVs in nine bat species from six locations. Furthermore, using the Illumina platform, we obtained two representative full-length genomes of the bat CoVs, namely TyRo-CoV-162275 and TyRo-CoV-162269. Sequence analysis showed that TyRo-CoV-162275 shared the highest identity with Malayan pangolin (Manis javanica) HKU4-related coronaviruses (MjHKU4r-CoVs) from Guangxi Province, whereas TyRo-CoV-162269 was closely related to HKU33-CoV discovered in a greater bamboo bat (Tylonycteris robustula) from Guizhou Province. Notably, TyRo-CoV-162275 has a putative furin protease cleavage site in its S protein and is likely to utilize human dipeptidyl peptidase-4 (hDPP4) as a cell-entry receptor, similar to MERS-CoV. To the best of our knowledge, this is the first report of a bat HKU4r-CoV strain containing a furin protease cleavage site. These findings expand our understanding of coronavirus geographic and host distributions.
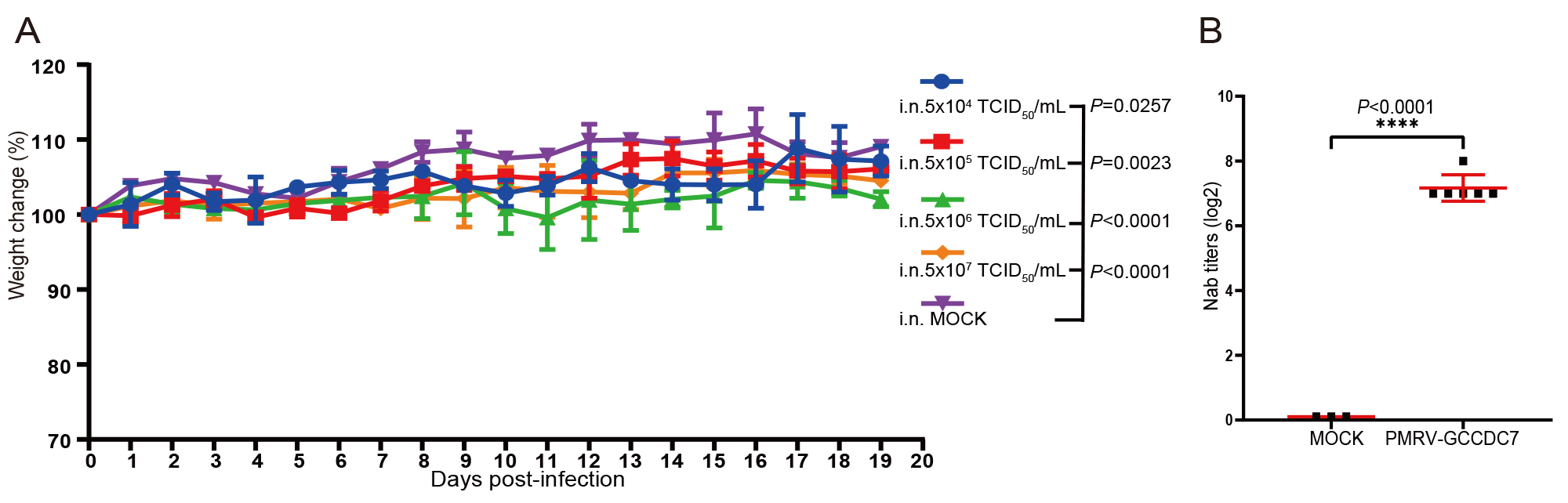
Emerging and re-emerging viruses from wild animals have seriously threatened the health of humans and domesticated animals in recent years. Herein, we isolated a new mammalian orthoreovirus (MRV), Pika/MRV/GCCDC7/2019 (PMRV-GCCDC7), in the Qinghai-Tibet Plateau wild pika (Ochotona curzoniae). Though the PMRV-GCCDC7 shows features of a typical reovirus with ten gene segments arranged in 3:3:4 in length, the virus belongs to an independent evolutionary branch compared to other MRVs based on phylogenetic tree analysis. The results of cellular susceptibility, species tropism, and replication kinetics of PMRV-GCCDC7 indicated the virus could infect four human cell lines (A549, Huh7, HCT, and LoVo) and six non-human cell lines, including Vero-E6, LLC-MK2, BHK-21, N2a, MDCK, and RfKT cell, derived from diverse mammals, i.e. monkey, mice, canine and bat, which revealed the potential of PMRV-GCCDC7 to infect a variety of hosts. Infection of BALB/c mice with PMRV-GCCDC7 via intranasal inoculation led to relative weight loss, lung tissue damage and inflammation with the increase of virus titer, but no serious respiratory symptoms and death occurred. The characterization of the new reovirus from a plateau-based wild animal has expanded our knowledge of the host range of MRV and provided insight into its risk of trans-species transmission and zoonotic diseases.
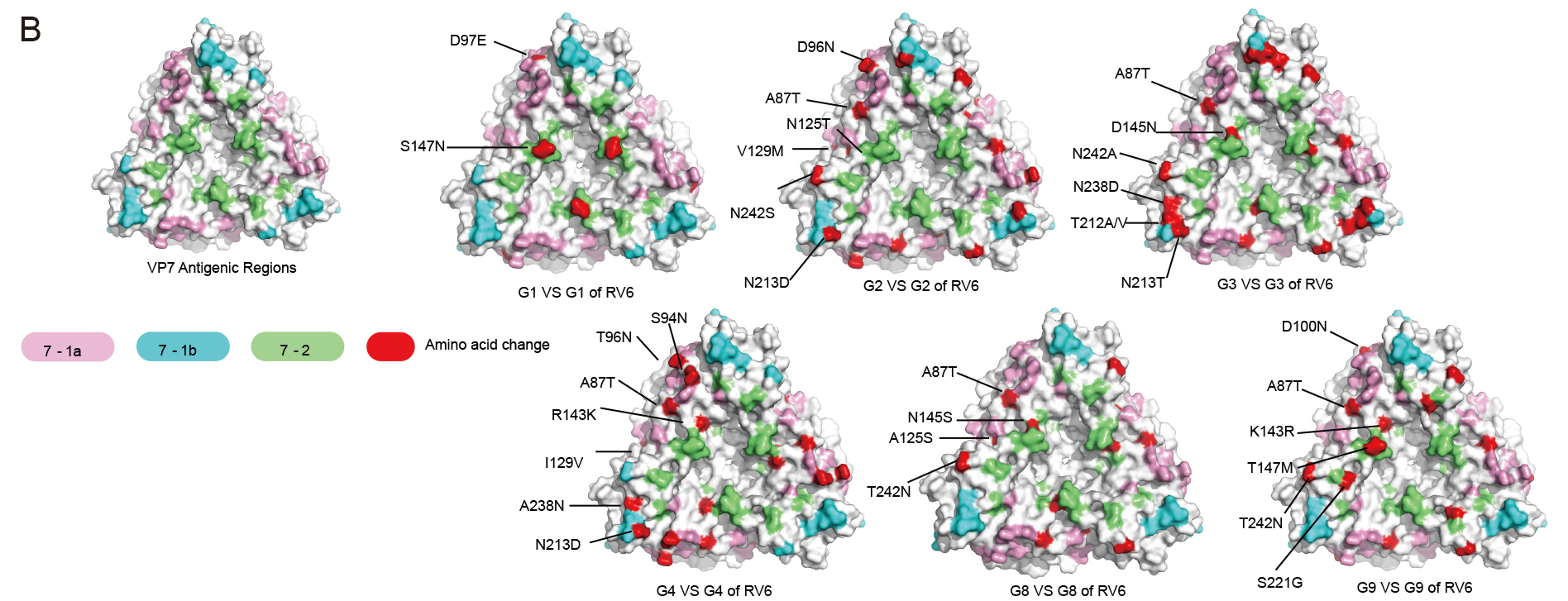
The oral hexavalent live human-bovine reassortant rotavirus vaccine (RV6) developed by Wuhan Institute of Biological Products Co., Ltd (WIBP) has finished a randomized, placebo-controlled phase III clinical trial in four provinces of China in 2021. The trail demonstrated that RV6 has a high vaccine efficacy against the prevalent strains and is safe for use in infants. During the phase III clinical trial (2019–2021), 200 rotavirus-positive fecal samples from children with RV gastroenteritis (RVGE) were further studied. Using reverse transcription-polymerase chain reaction and high-throughput sequencing, VP7 and VP4 sequences were obtained and their genetic characteristics, as well as the differences in antigenic epitopes of VP7, were analyzed in detail. Seven rotavirus genotypes were identified. The predominant rotavirus genotype was G9P [8] (77.0%), followed by prevalent strains G8P [8] (8.0%), G3P [8] (3.5%), G3P [9] (1.5%), G1P [8] (1.0%), G2P [4] (1.0%), and G4P [6] (1.0%). The amino acid sequence identities of G1, G2, G3, G4, G8, and G9 genotypes of isolates compared to the vaccine strains were 98.8%, 98.2%–99.7%, 88.4%–99.4%, 98.2%, 94.2%–100%, and 93.9%–100%, respectively. Notably, the vaccine strains exhibited high similarity in amino acid sequence, with only minor differences in antigenic epitopes compared to the Chinese endemic strains. This supports the potential application of the vaccine in preventing diseases caused by rotaviruses.
The NS5A non-structural protein of classical swine fever virus (CSFV) is a multifunctional protein involved in viral genomic replication, protein translation, assembly of infectious virus particles, and regulation of cellular signaling pathways. Previous report showed that NS5A inhibited nuclear factor kappa B (NF-κB) signaling induced by poly(I:C); however, the mechanism involved has not been elucidated. Here, we reported that NS5A directly interacted with NF-κB essential modulator (NEMO), a regulatory subunit of the IκB kinase (IKK) complex, to inhibit the NF-κB signaling pathway. Further investigations showed that the zinc finger domain of NEMO and the aa 126–250 segment of NS5A are essential for the interaction between NEMO and NS5A. Mechanistic analysis revealed that NS5A mediated the proteasomal degradation of NEMO. Ubiquitination assay showed that NS5A induced the K27-linked but not the K48-linked polyubiquitination of NEMO for proteasomal degradation. In addition, NS5A blocked the K63-linked polyubiquitination of NEMO, thus inhibiting IKK phosphorylation, IκBα degradation, and NF-κB activation. These findings revealed a novel mechanism by which CSFV inhibits host innate immunity, which might guide the drug design against CSFV in the future.
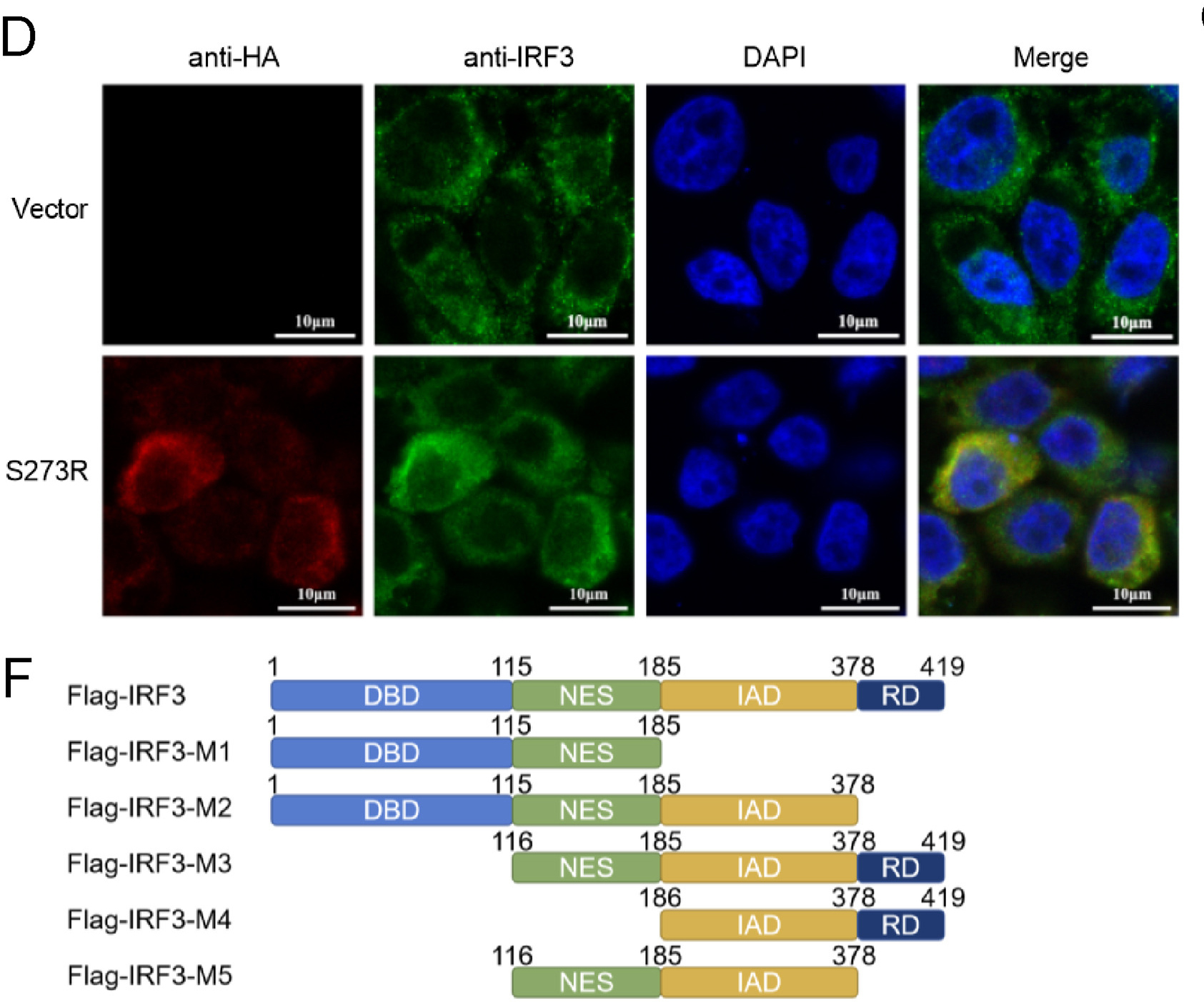
African swine fever (ASF) is originally reported in East Africa as an acute hemorrhagic fever. African swine fever virus (ASFV) is a giant and complex DNA virus with icosahedral structure and encodes a variety of virulence factors to resist host innate immune response. S273R protein (pS273R), as a SUMO-1 specific cysteine protease, can affect viral packaging by cutting polymeric proteins. In this study, we found that pS273R was an important antagonistic viral factor that suppressed cGAS-STING-mediated type I interferon (IFN-I) production. A detailed analysis showed that pS273R inhibited IFN-I production by interacting with interferon regulatory factor 3 (IRF3). Subsequently, we showed that pS273R disrupted the association between TBK1 and IRF3, leading to the repressed IRF3 phosphorylation and dimerization. Deletion and point mutation analysis verified that pS273R impaired IFN-I production independent of its cysteine protease activity. These findings will help us further understand ASFV pathogenesis.
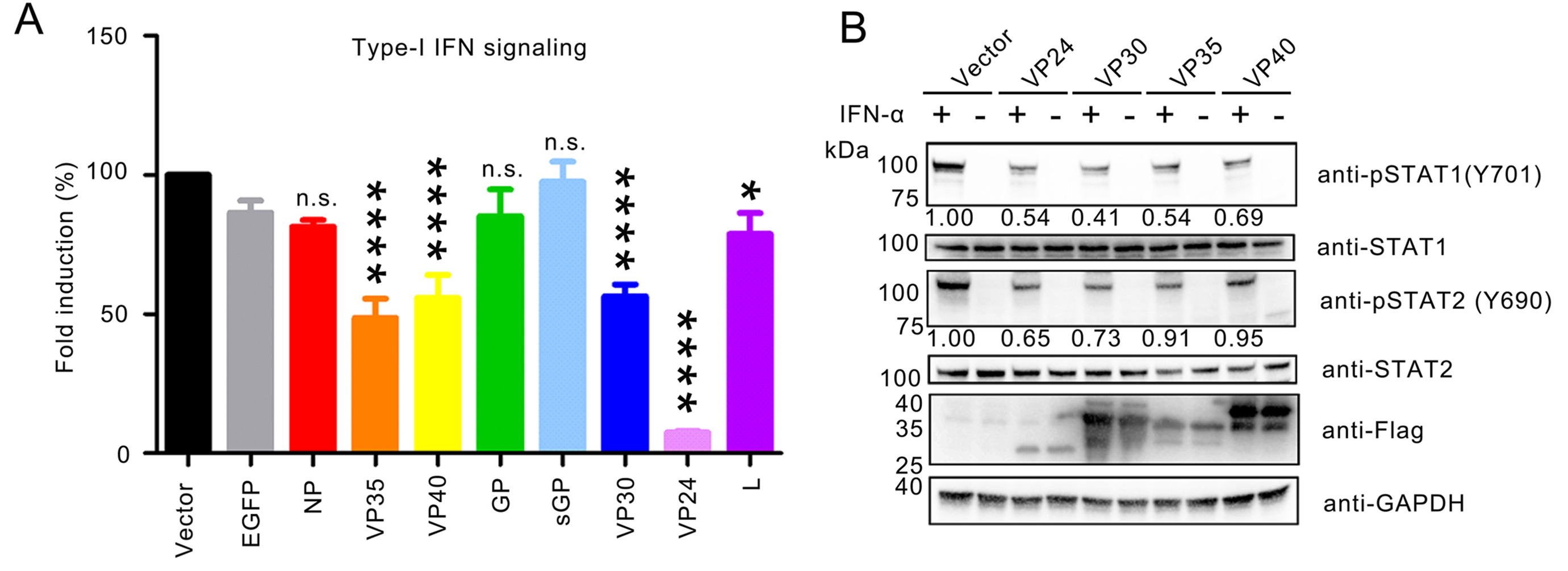
As one of the deadliest viruses, Ebola virus (EBOV) causes lethal hemorrhagic fevers in humans and nonhuman primates. The suppression of innate immunity leads to robust systemic virus replication of EBOV, leading to enhanced transmission. However, the mechanism of EBOV-host interaction is not fully understood. Here, we identified multiple dysregulated genes in early stage of EBOV infection through transcriptomic analysis, which are highly clustered to Jak-STAT signaling. EBOV VP35 and VP30 were found to inhibit type I interferon (IFN) signaling. Moreover, exogenous expression of VP35 blocks the phosphorylation of endogenous STAT1, and suppresses nuclear translocation of STAT1. Using serial truncated mutations of VP35, N-terminal 1–220 amino acid residues of VP35 were identified to be essential for blocking on type I IFN signaling. Remarkably, VP35 of EBOV suppresses type I IFN signaling more efficiently than those of Bundibugyo virus (BDBV) and Marburg virus (MARV), resulting in stable replication to facilitate the pathogenesis. Altogether, this study enriches understanding on EBOV evasion of innate immune response, and provides insights into the interplay between filoviruses and host.
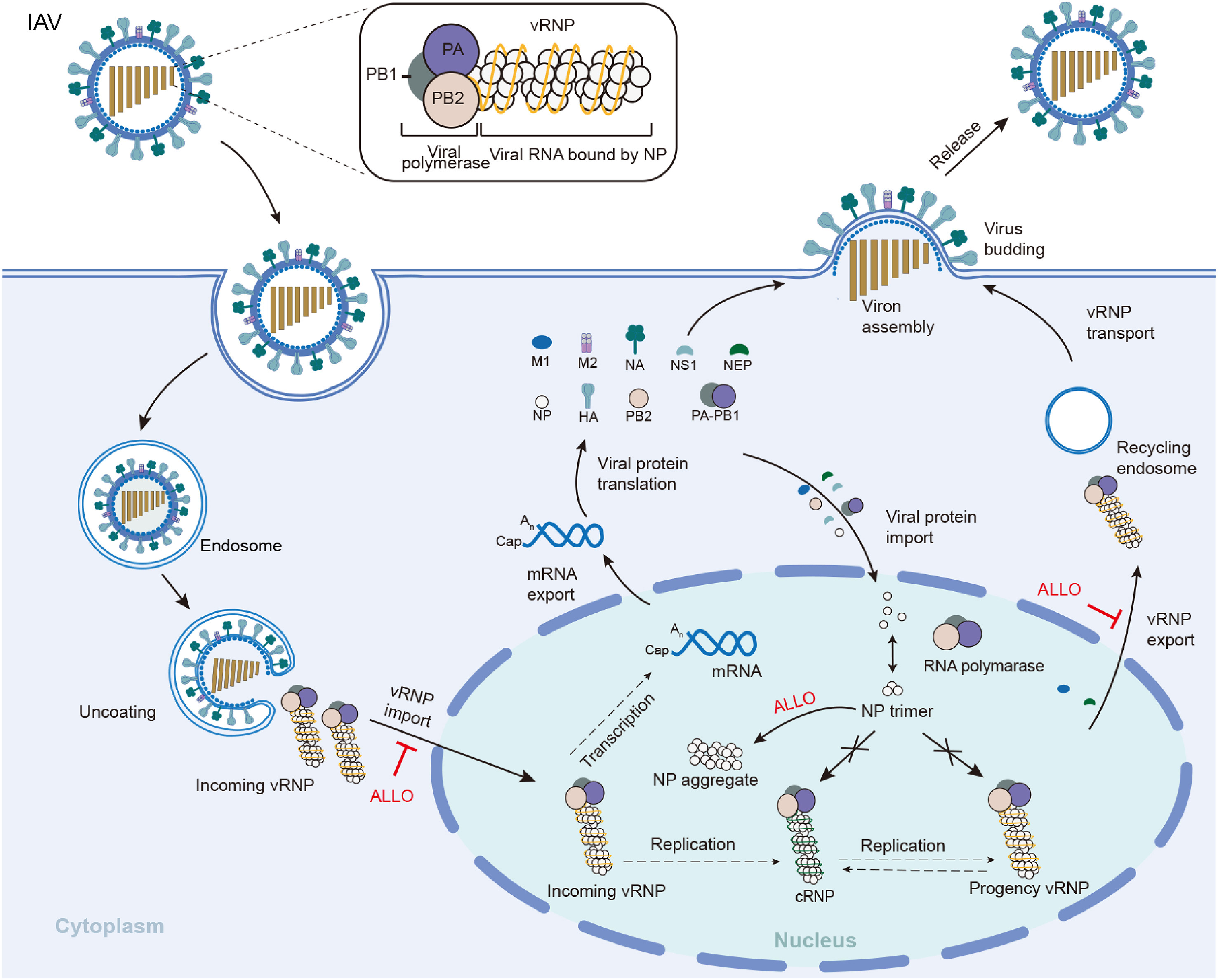
Influenza A virus (IAV) poses a global public health concern and remains an imminent threat to human health. Emerging antiviral resistance to the currently approved influenza drugs emphasizes the urgent need for new therapeutic entities against IAV. Allopregnanolone (ALLO) is a natural product that has been approved as an antidepressant drug. In the present study, we repurposed ALLO as a novel inhibitor against IAVs. Mechanistic studies demonstrated that ALLO inhibited virus replication by interfering with the nucleus translocation of viral nucleoprotein (NP). In addition, ALLO showed significant synergistic activity with compound 16, a hemagglutinin inhibitor of IAVs. In summary, we have identified ALLO as a novel influenza virus inhibitor targeting NP, providing a promising candidate that deserves further investigation as a useful anti-influenza strategy in the future.
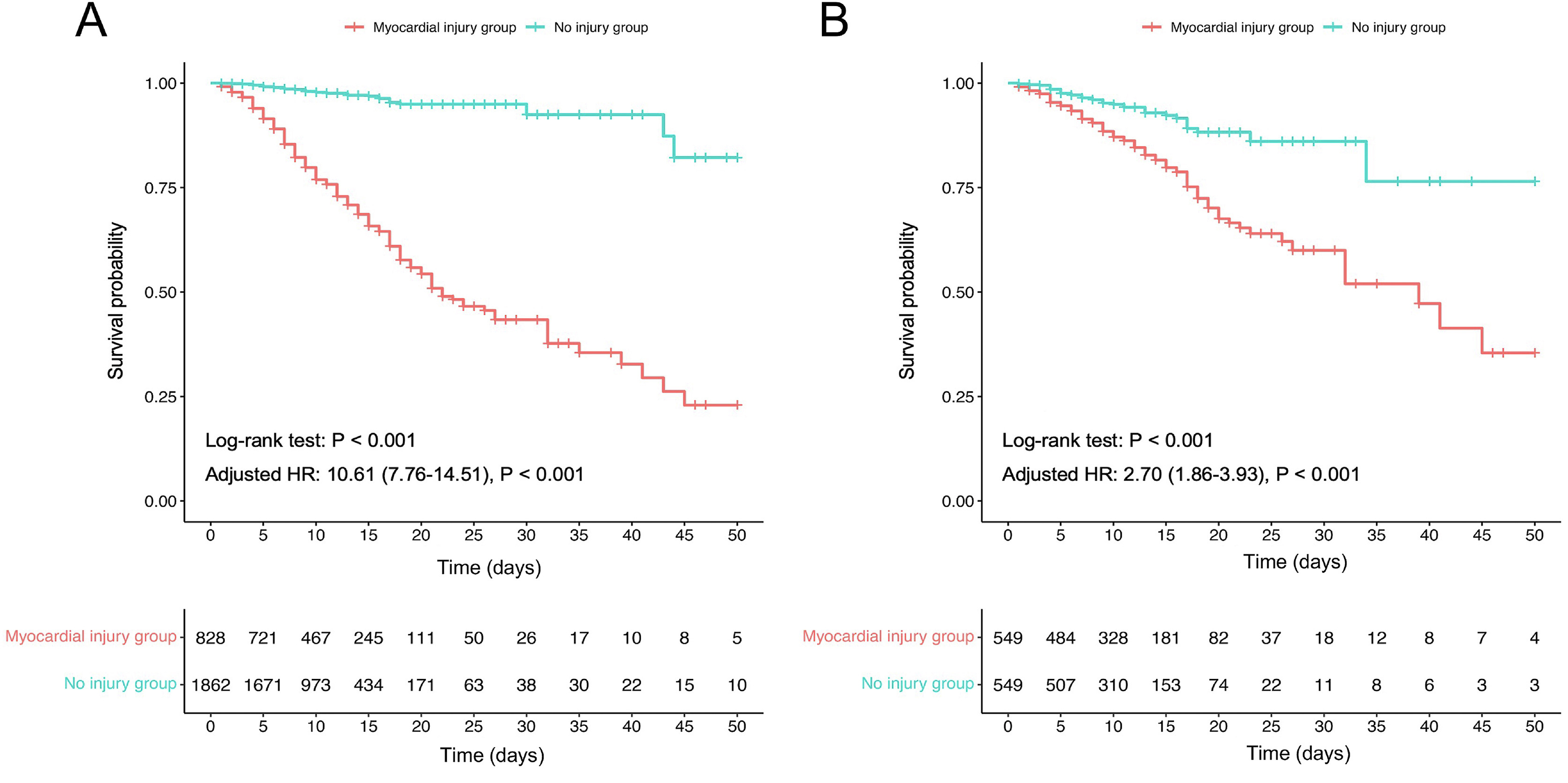
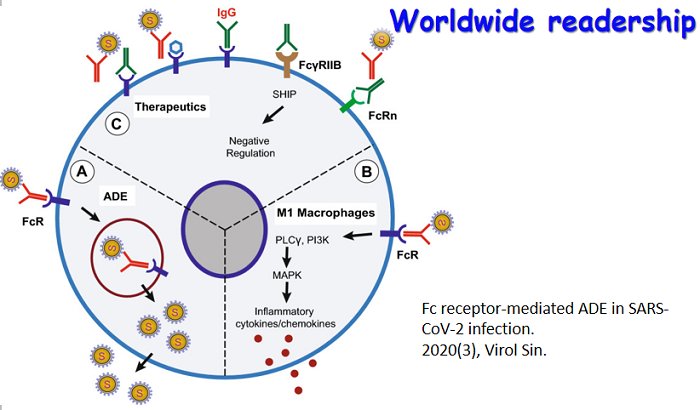
Myocardial injury is one of the most common comorbidity in SARS-CoV-2 infected patients, and has poor prognosis. However, the incidence of myocardial injury in patients with SARS-CoV-2 infection has not been sufficiently investigated during the Omicron wave. We conducted a retrospective study of 2690 patients with confirmed SARS-CoV-2 Omicron infection from Tongji Hospital. The results indicated that the myocardial injury accounted for 30.8% of the total patients with SARS-CoV-2 infection and was associated with higher in-hospital mortality than those without injury before and after propensity score matching (PSM) [adjusted hazard ratio (HR), 10.61; 95% confidence interval (CI), 7.76–14.51; P < 0.001; adjusted HR, 2.70; 95% CI, 1.86–3.93; P < 0.001; respectively]. Further, the levels of cytokines (IL-1β, IL-6, IL-10, and TNF-α) in patients with myocardial injury were higher than those without injury, and the higher levels of cytokines in the myocardial injury group were associated with increased mortality. Administration of angiotensin-converting enzyme inhibitors or angiotensin receptor blockers (ACEI/ARB) could significantly reduce the mortality in patients with myocardial injury (adjusted HR, 0.52; 95% CI, 0.38–0.71; P < 0.001). Additionally, the level of angiotensin II increased in patients with SARS-CoV-2 infection was even higher in myocardial injury group compared to those without injury. Collectively, the study summarized the clinical characteristic and outcome of SARS-CoV-2 infected patients with myocardial injury during the Omicron wave in China, and validated the protective role of ACEI/ARB in improving the survival of those with myocardial injury.
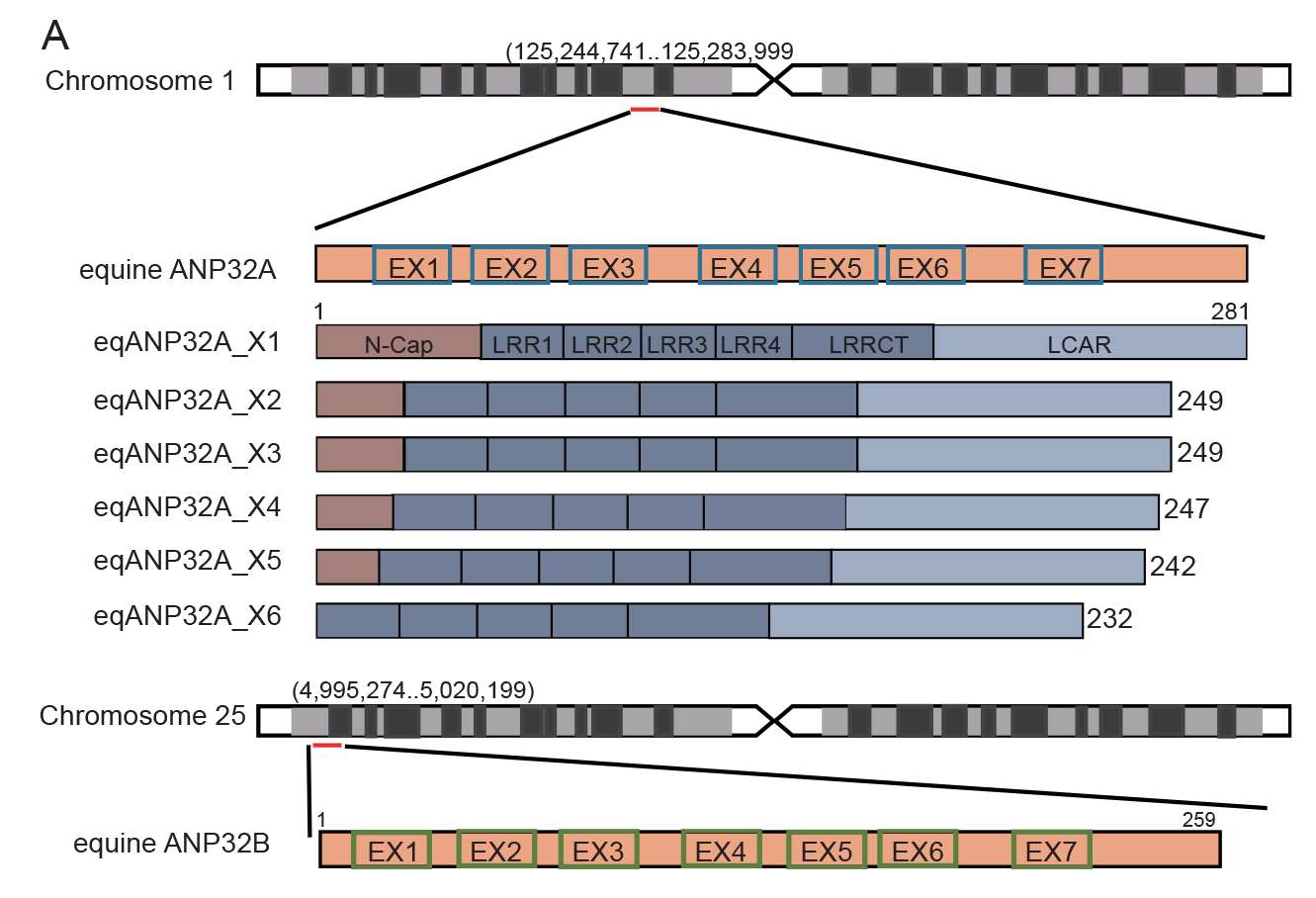
Host ANP32 family proteins are crucial for maintaining the activity of influenza RNA polymerase and play an important role in the cross-species transmission of influenza viruses. To date, the molecular properties of equine ANP32 (eqANP32) protein are poorly understood, particularly the mechanisms that affect equine influenza virus (EIV) RNA polymerase activity. Here, we found that there are six alternative splicing variants of equine ANP32A (eqANP32A) with different levels of expression. Further studies showed that these six splicing variants of eqANP32A supported the activity of EIV RNA polymerase to varying degrees, with the variant eqANP32A_X2 having the highest expression abundance and exhibiting the highest support of polymerase activity. Sequence analysis demonstrated that the differences in the N-Cap regions of the six splicing variants significantly affected their N-terminal conformation, but did not affect their ability to bind RNA polymerase. We also demonstrated that there is only one transcript of eqANP32B, and that this transcript showed only very low support to the EIV RNA polymerase. This functional defect in eqANP32B is caused by the sequence of the 110–259 amino acids at its C-terminus. Our results indicated that it is the eqANP32A_X2 protein that mainly determines the efficiency of the EIV replication in horses. In conclusion, our study parsed the molecular properties of eqANP32 family proteins and revealed the sequence features of eqANP32A and eqANP32B, suggesting for the first time that the N-Cap region of ANP32A protein also plays an important role in supporting the activity of the influenza virus polymerase.
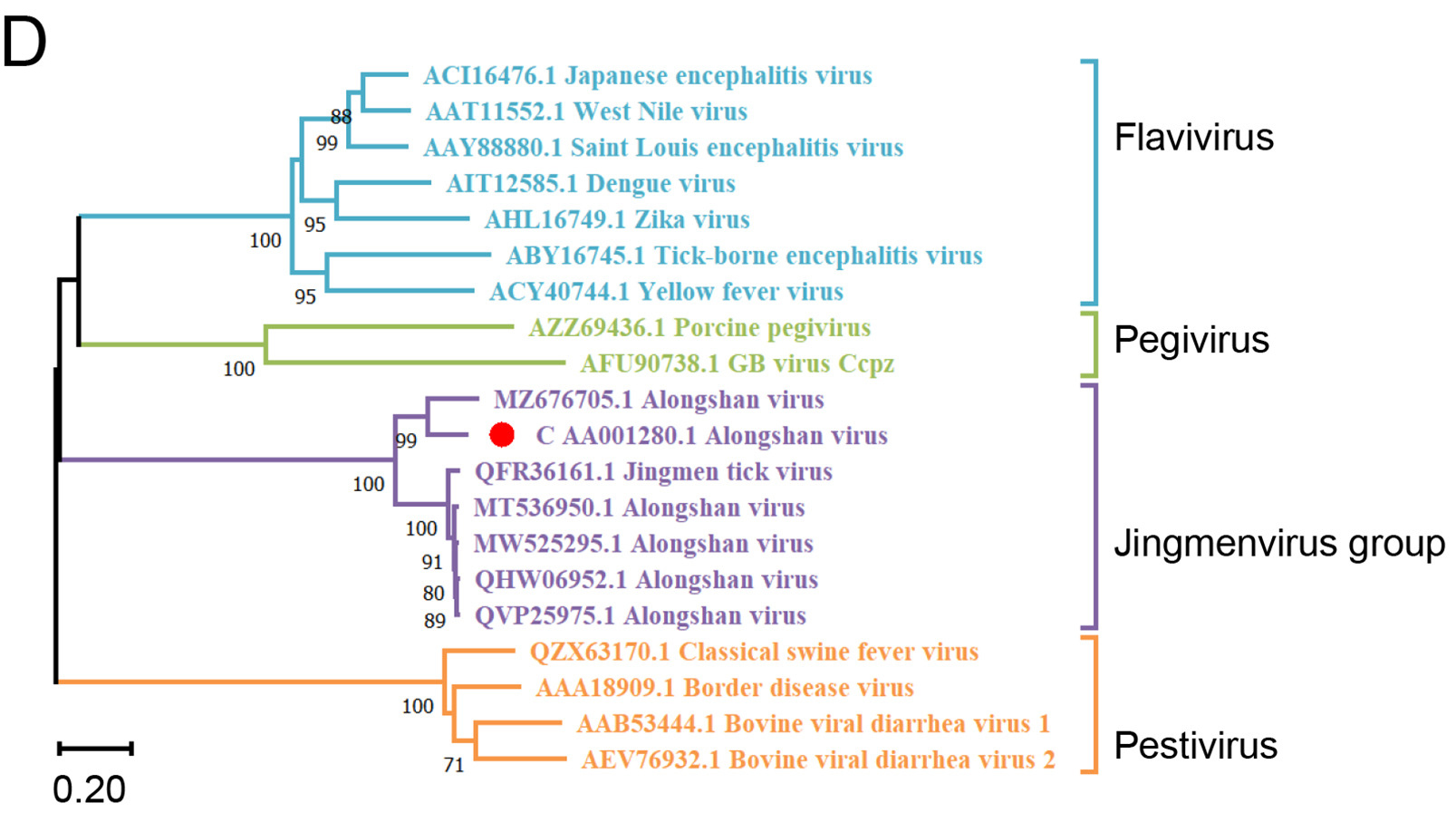
Highlights 1. This study identified eleven tick-borne viruses in Liaoning Province and Inner Mongolia, China, which greatly enrich the diversity of tick-borne virus. 2. Tacheng tick virus 2 is for the first time detected outside Xinjiang and in a novel tick species D. niveus. 3. The Alongshan virus and Tacheng tick virus 2 identified in this study can be considered as novel species.
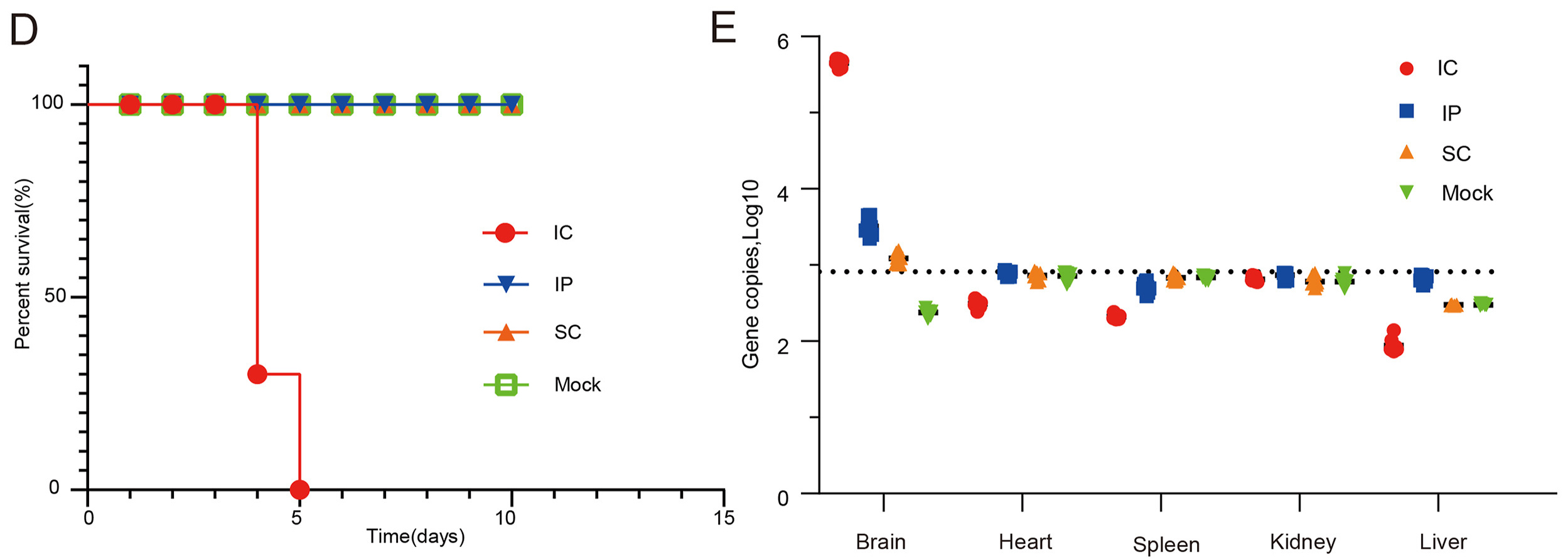
Highlights 1. A novel Akabane virus strain LK07 was isolated from cattle. 2. Here, we firstly reported the identification and isolation of the Akabane virus sub-lineage genogroup Ib in China. 3. The suckling mice showed clinical symptoms after intracerebrally infected with LK07 strain. 4. Viral antigen was detected in brain of mice infected with LK07 strain via IC, IP and SC routes.
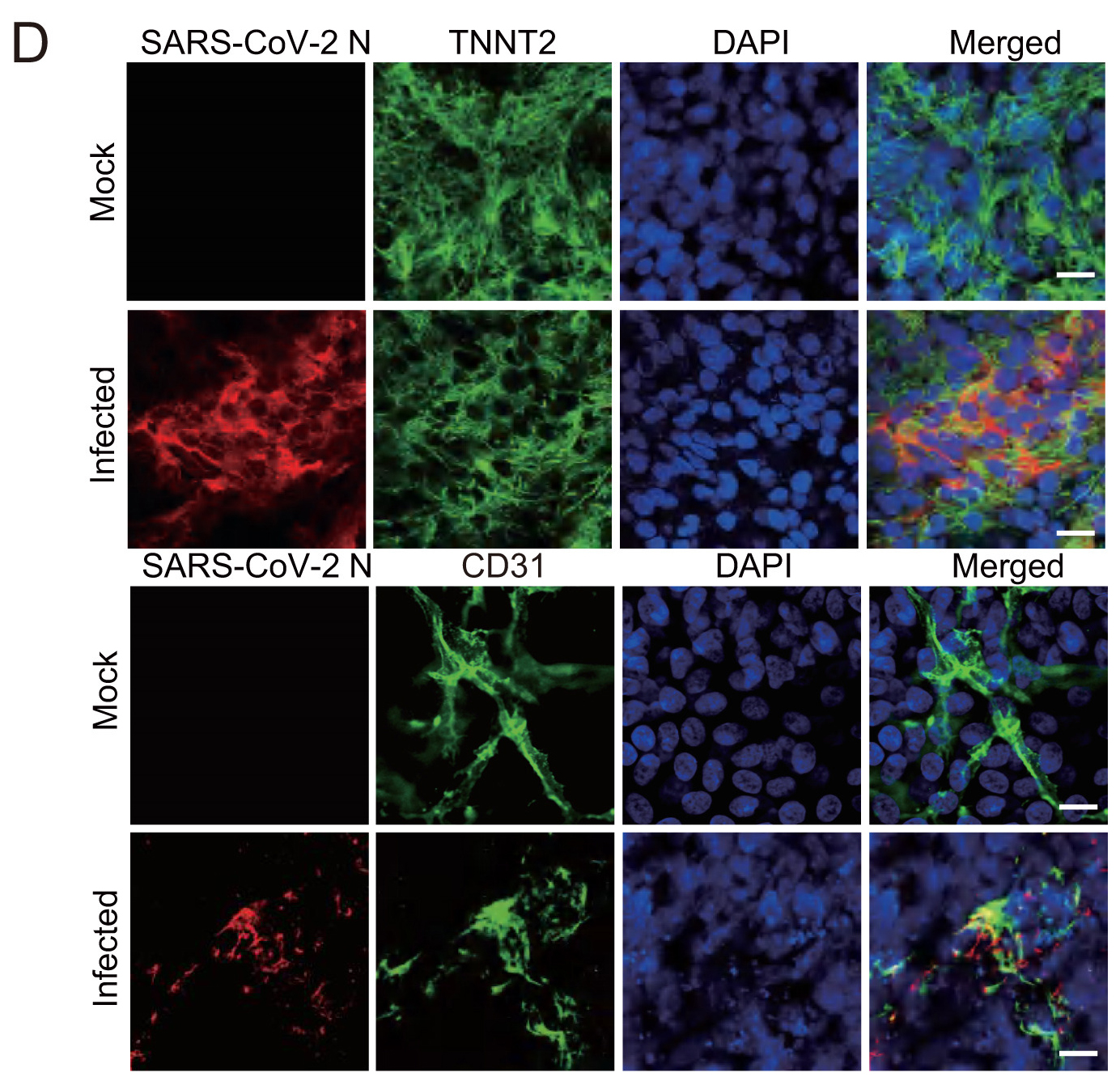
Highlights 1. Establishment of 3D cardiac organoids composed of cardiomyocyte and endothelial layers. 2. SARS-CoV-2 infection causes multi-lineage cardiac injuries. 3. Cardiovascular health should be greatly concerned in COVID-19 patients.
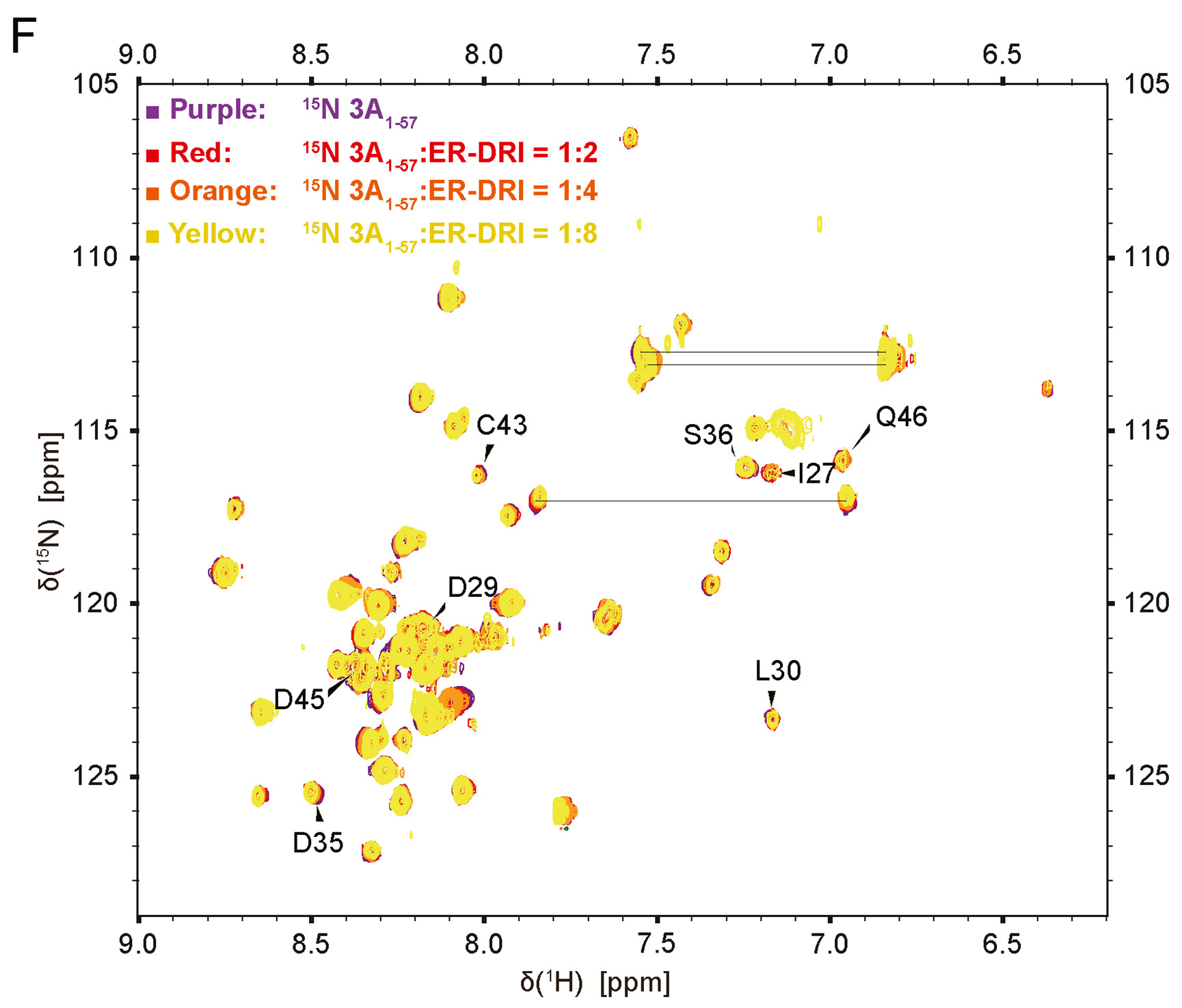
Highlights 1. Our results disclosed a dihelical structure of Enterovirus A71 3A1-57 protein in apo form. 2. We depicted rigid helices and a unique flexible C-terminus for apo-form 3A1-57. 3. This study revealed a competitive binding-based molecular mechanism underlying inhibition of dimeric 3A by ER-DRI.