In 2018–2019, both incidence of pediatric HAdV infection and severe illness cases substantially increased in the southern regions of China. To analyze the characteristics and risk factors of this severe HAdV epidemic, Liu et al. performed a comprehensive investigation on the clinical characteristics of patients with severe HAdV infection, HAdV type distribution, viral genome features, and in vitro viral proliferation and cytopathogenesis properties. This study would provide important reference data for the treatment, control, and prevention of HAdV infection. The cover shows that kids with HAdV-7 infection, comorbid disease and HAdV co-infection may progress to severe illness (kindly provided by Wenkuan Liu and Rong Zhou). See 331-340 pages for details.
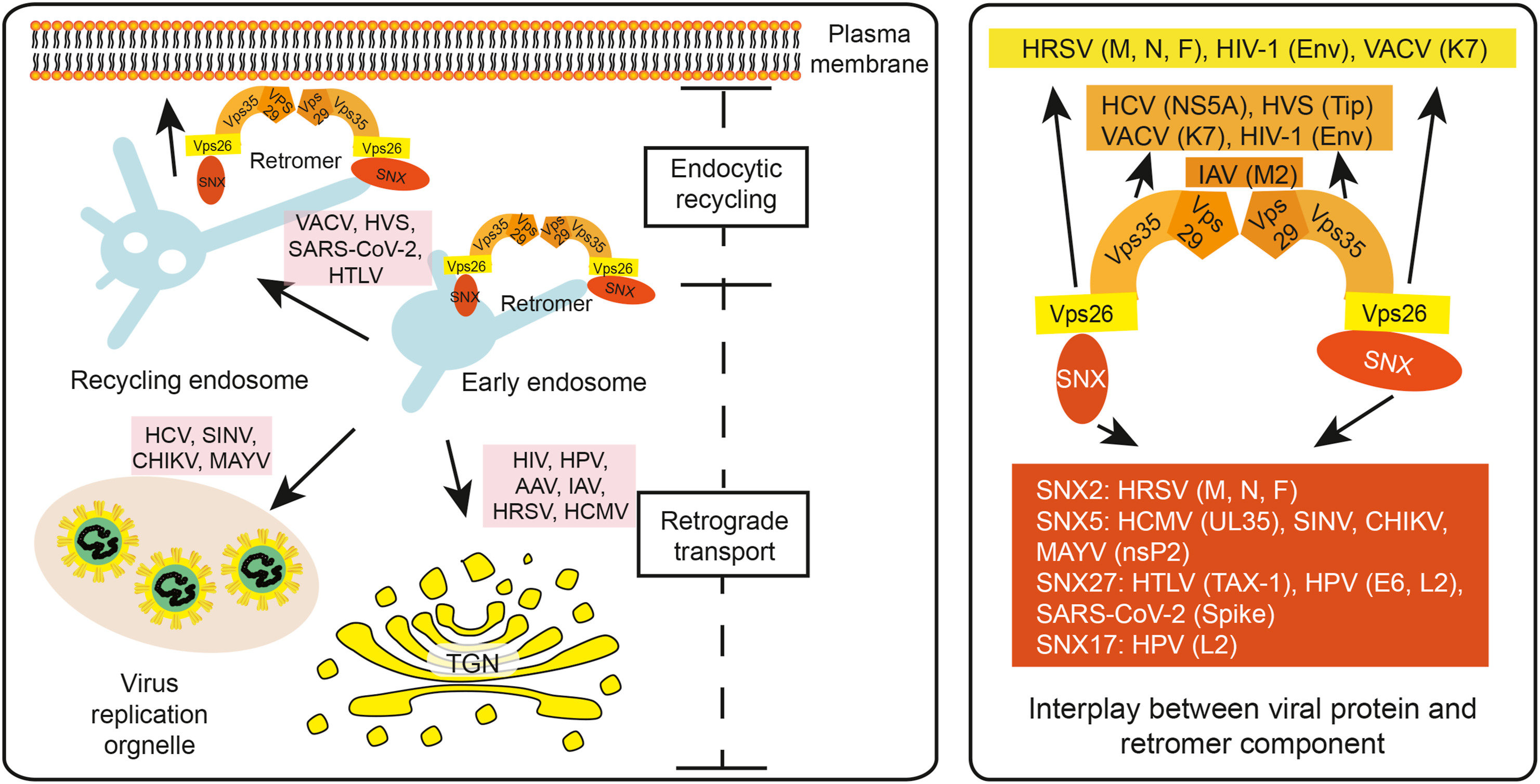
Yue Lu, Ping He, Yuxuan Zhang, Yongwen Ren and Leiliang Zhang. The emerging roles of retromer and sorting nexins in the life cycle of viruses[J]. Virologica Sinica, 2022, 37(3): 321-330. doi: 10.1016/j.virs.2022.04.014.
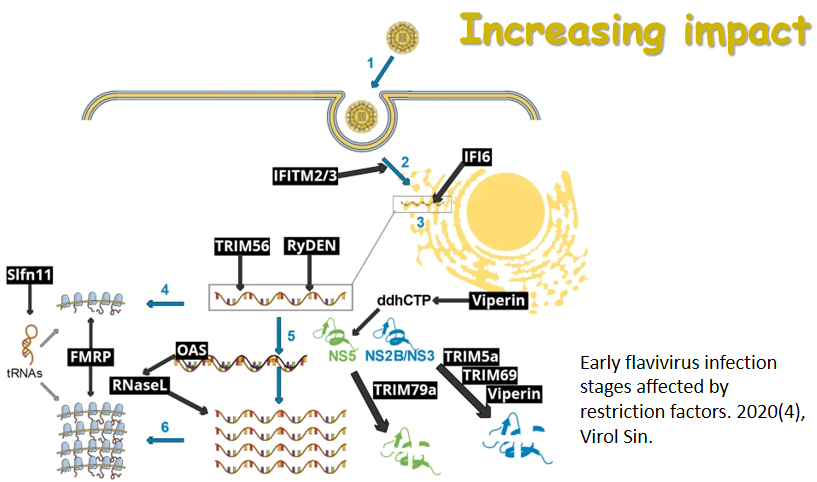
Retromer and sorting nexins (SNXs) transport cargoes from endosomes to the trans-Golgi network or plasma membrane. Recent studies have unveiled the emerging roles for retromer and SNXs in the life cycle of viruses, including members of Coronaviridae, Flaviviridae and Retroviridae. Key components of retromer/SNXs, such as Vps35, Vps26, SNX5 and SNX27, can affect multiple steps of the viral life cycle, including facilitating the entry of viruses into cells, participating in viral replication, and promoting the assembly of virions. Here we present a comprehensive updated review on the interplay between retromer/SNXs and virus, which will shed mechanistic insights into controlling virus infection.
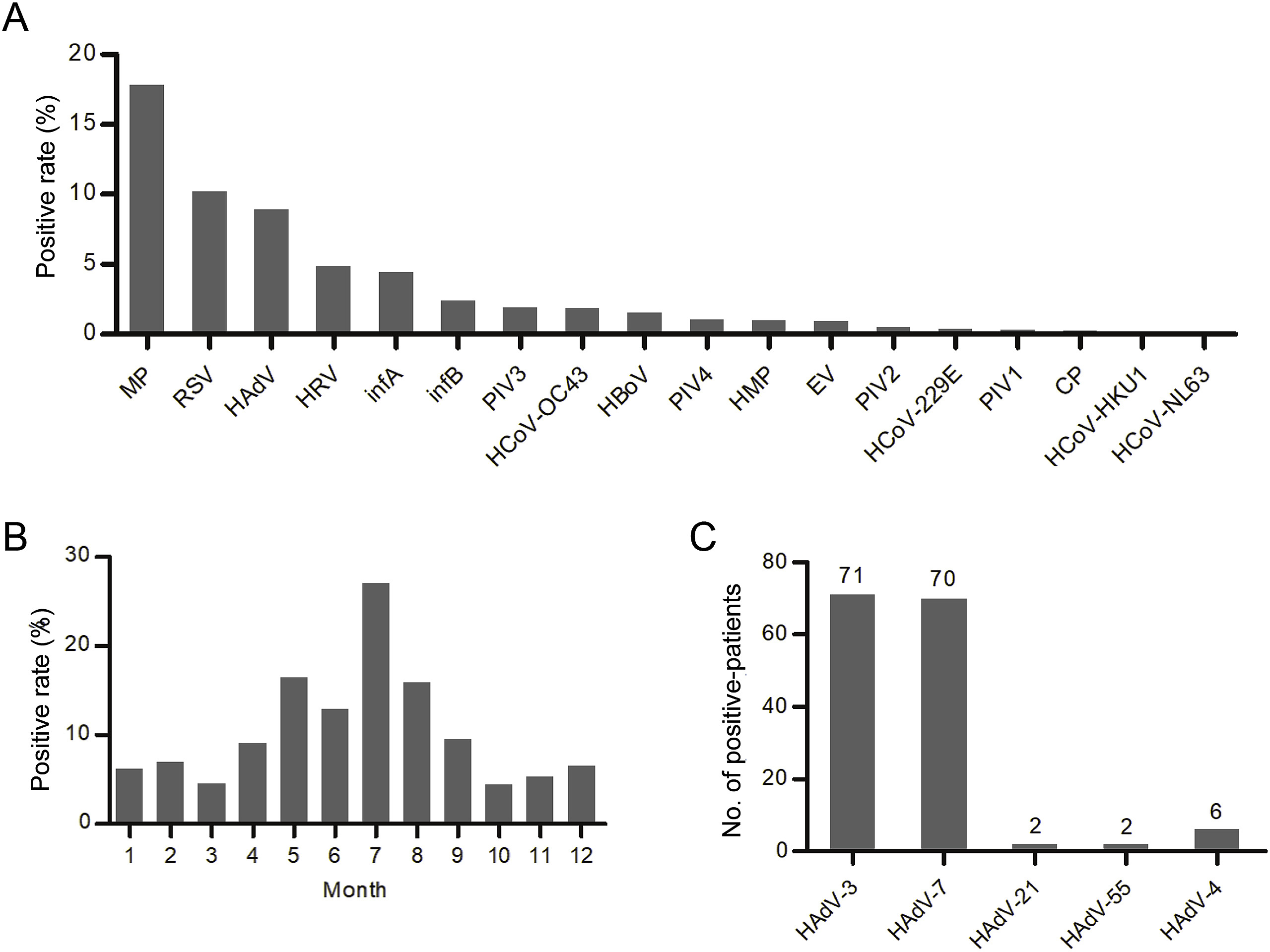
Wenkuan Liu, Shuyan Qiu, Li Zhang, Hongkai Wu, Xingui Tian, Xiao Li, Duo Xu, Jing Dai, Shujun Gu, Qian Liu, Dehui Chen and Rong Zhou. Analysis of severe human adenovirus infection outbreak in Guangdong Province, southern China in 2019[J]. Virologica Sinica, 2022, 37(3): 331-340. doi: 10.1016/j.virs.2022.01.010.
During 2018-2019, a severe human adenovirus (HAdV) infection outbreak occurred in southern China. Here, we screened 18 respiratory pathogens in 1704 children (≤ 14 years old) hospitalized with acute respiratory illness in Guangzhou, China, in 2019. In total, 151 patients had positive HAdV test results; 34.4% (52/151) of them exhibited severe illness. HAdV infection occurred throughout the year, with a peak in summer. The median patient age was 3.0 (interquartile range:1.1-5.0) years. Patients with severe HAdV infection exhibited increases in 12 clinical indexes (P ≤ 0.019) and decreases in four indexes (P ≤ 0.007), compared with patients exhibiting nonsevere infection. No significant differences were found in age or sex distribution according to HAdV infection severity (P > 0.05); however, the distributions of comorbid disease and HAdV co-infection differed according to HAdV infection severity (P < 0.05). The main epidemic types were HAdV-3 (47.0%, 71/151) and HAdV-7 (46.4%, 70/151). However, the severe illness rate was significantly higher in patients with HAdV-7 (51.4%) than in patients with HAdV-3 (19.7%) and other types of HAdV (20%) (P < 0.001). Sequencing analysis of genomes/capsid genes of 13 HAdV-7 isolates revealed high similarity to previous Chinese isolates. A representative HAdV-7 isolate exhibited a similar proliferation curve to the curve described for the epidemic HAdV-3 strain Guangzhou01 (accession no. DQ099432) (P > 0.05); the HAdV-7 isolate exhibited stronger virulence and infectivity, compared with HAdV-3 (P < 0.001). Overall, comorbid disease, HAdV co-infection, and high virulence and infectivity of HAdV-7 were critical risk factors for severe HAdV infection; these data can facilitate treatment, control, and prevention of HAdV infection.
Lei Yang, Lingqian Tian, Leshan Li, Qiuhong Liu, Xiang Guo, Yuan Zhou, Rongjuan Pei, Xinwen Chen and Yun Wang. Efficient assembly of a large fragment of monkeypox virus genome as a qPCR template using dual-selection based transformation-associated recombination[J]. Virologica Sinica, 2022, 37(3): 341-347. doi: 10.1016/j.virs.2022.02.009.
Transformation-associated recombination (TAR) has been widely used to assemble large DNA constructs. One of the significant obstacles hindering assembly efficiency is the presence of error-prone DNA repair pathways in yeast, which results in vector backbone recircularization or illegitimate recombination products. To increase TAR assembly efficiency, we prepared a dual-selective TAR vector, pGFCS, by adding a PADH1-URA3 cassette to a previously described yeast-bacteria shuttle vector, pGF, harboring a PHIS3-HIS3 cassette as a positive selection marker. This new cassette works as a negative selection marker to ensure that yeast harboring a recircularized vector cannot propagate in the presence of 5-fluoroorotic acid. To prevent pGFCS bearing ura3 from recombining with endogenous ura3-52 in the yeast genome, a highly transformable Saccharomyces cerevisiae strain, VL6-48B, was prepared by chromosomal substitution of ura3-52 with a transgene conferring resistance to blasticidin. A 55-kb genomic fragment of monkeypox virus encompassing primary detection targets for quantitative PCR was assembled by TAR using pGFCS in VL6-48B. The pGFCS-mediated TAR assembly showed a zero rate of vector recircularization and an average correct assembly yield of 79% indicating that the dual-selection strategy provides an efficient approach to optimizing TAR assembly.
Tongwei Ren, Xiangling Min, Qingrong Mo, Yuxu Wang, Hao Wang, Ying Chen, Kang Ouyang, Weijian Huang and Zuzhang Wei. Construction and characterization of a full-length infectious clone of Getah virus in vivo[J]. Virologica Sinica, 2022, 37(3): 348-357. doi: 10.1016/j.virs.2022.03.007.
Getah virus (GETV) is a mosquito-borne virus of the genus Alphavirus in the family Togaviridae and, in recent years, it has caused several outbreaks in animals. The molecular basis for GETV pathogenicity is not well understood. Therefore, a reverse genetic system of GETV is needed to produce genetically modified viruses for the study of the viral replication and its pathogenic mechanism. Here, we generated a CMV-driven infectious cDNA clone based on a previously isolated GETV strain, GX201808 (pGETV-GX). Transfection of pGETV-GX into BHK- 21 cells resulted in the recovery of a recombinant virus (rGETV-GX) which showed similar growth characteristics to its parental virus. Then three-day-old mice were experimentally infected with either the parental or recombinant virus. The recombinant virus showed milder pathogenicity than the parental virus in the mice. Based on the established CMV-driven cDNA clone, subgenomic promoter and two restriction enzyme sites (BamHI and EcoRI) were introduced into the region between E1 protein and 30UTR. Then the green fluorescent protein (GFP), red fluorescent protein (RFP) and improved light-oxygen-voltage (iLOV) genes were inserted into the restriction enzyme sites. Transfection of the constructs carrying the reporter genes into BHK-21 cells proved the rescue of the recombinant reporter viruses. Taken together, the establishment of a reverse genetic system for GETV provides a valuable tool for the study of the virus life cycle, and to aid the development of genetically engineered GETVs as vectors for foreign gene expression.
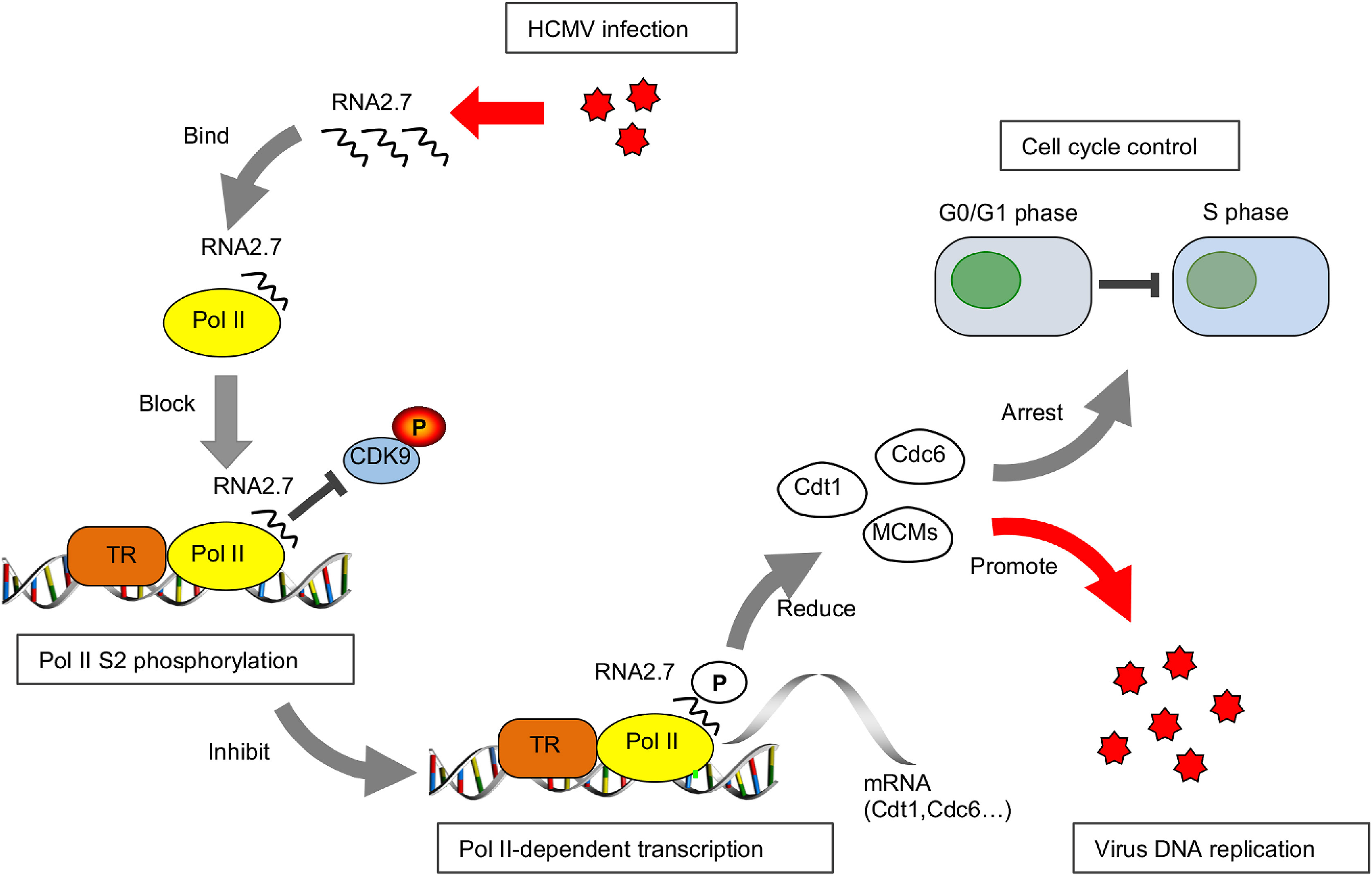
Yujing Huang, Xin Guo, Jing Zhang, Jianming Li, Mingyi Xu, Qing Wang, Zhongyang Liu, Yanping Ma, Ying Qi and Qiang Ruan. Human cytomegalovirus RNA2.7 inhibits RNA polymerase II (Pol II) Serine-2 phosphorylation by reducing the interaction between Pol II and phosphorylated cyclin-dependent kinase 9 (pCDK9)[J]. Virologica Sinica, 2022, 37(3): 358-369. doi: 10.1016/j.virs.2022.02.011.
Human cytomegalovirus (HCMV) is a ubiquitous pathogen belongs to betaherpesvirus subfamily. RNA2.7 is a highly conserved long non-coding RNA accounting for more than 20% of total viral transcripts. In our study, functions of HCMV RNA2.7 were investigated by comparison of host cellular transcriptomes between cells infected with HCMV clinical strain and RNA2.7 deleted mutant. It was demonstrated that RNA polymerase II (Pol II)- dependent host gene transcriptions were significantly activated when RNA2.7 was removed during infection. A 145 nt-in-length motif within RNA2.7 was identified to inhibit the phosphorylation of Pol II Serine-2 (Pol II S2) by reducing the interaction between Pol II and phosphorylated cyclin-dependent kinase 9 (pCDK9). Due to the loss of Pol II S2 phosphorylation, cellular DNA pre-replication complex (pre-RC) factors, including Cdt1 and Cdc6, were significantly decreased, which prevented more cells from entering into S phase and facilitated viral DNA replication. Our results provide new insights of HCMV RNA2.7 functions in regulation of host cellular transcription.
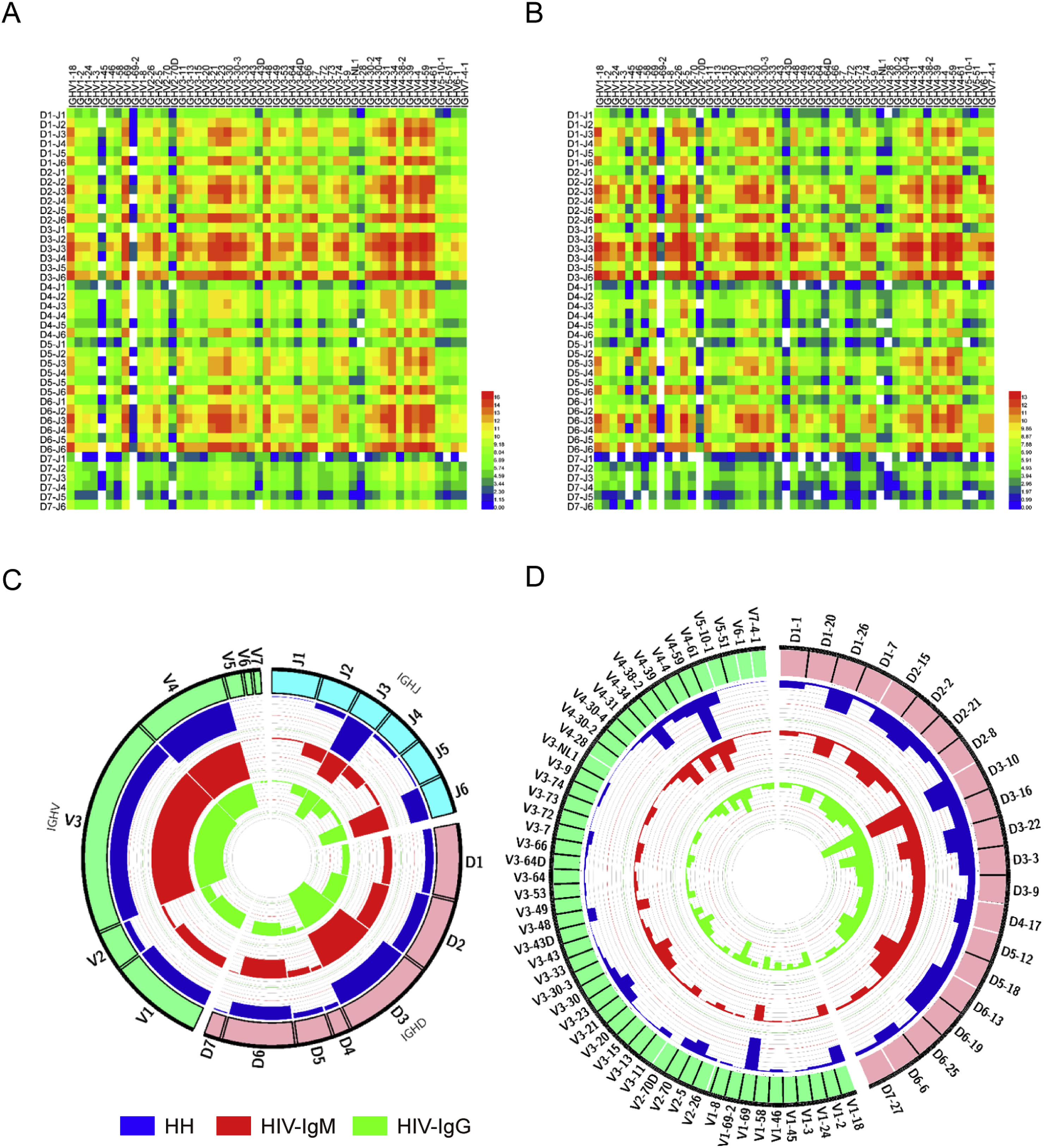
Xiaolong Tian, Binbin Hong, Xiaoyi Zhu, Desheng Kong, Yumei Wen, Yanling Wu, Liying Ma and Tianlei Ying. Characterization of human IgM and IgG repertoires in individuals with chronic HIV-1 infection[J]. Virologica Sinica, 2022, 37(3): 370-379. doi: 10.1016/j.virs.2022.02.010.
Advancements in high-throughput sequencing (HTS) of antibody repertoires (Ig-Seq) have unprecedentedly improved our ability to characterize the antibody repertoires on a large scale. However, currently, only a few studies explored the influence of chronic HIV-1 infection on human antibody repertoires and many of them reached contradictory conclusions, possibly limited by inadequate sequencing depth and throughput. To better understand how HIV-1 infection would impact humoral immune system, in this study, we systematically analyzed the differences between the IgM (HIV-IgM) and IgG (HIV-IgG) heavy chain repertoires of HIV-1 infected patients, as well as between antibody repertoires of HIV-1 patients and healthy donors (HH). Notably, the public unique clones accounted for only a negligible proportion between the HIV-IgM and HIV-IgG repertoires libraries, and the diversity of unique clones in HIV-IgG remarkably reduced. In aspect of somatic mutation rates of CDR1 and CDR2, the HIV-IgG repertoire was higher than HIV-IgM. Besides, the average length of CDR3 region in HIV-IgM was significant longer than that in the HH repertoire, presumably caused by the great number of novel VDJ rearrangement patterns, especially a massive use of IGHJ6. Moreover, some of the B cell clonotypes had numerous clones, and somatic variants were detected within the clonotype lineage in HIV-IgG, indicating HIV-1 neutralizing activities. The in-depth characterization of HIV-IgG and HIV-IgM repertoires enriches our knowledge in the profound effect of HIV-1 infection on human antibody repertoires and may have practical value for the discovery of therapeutic antibodies.
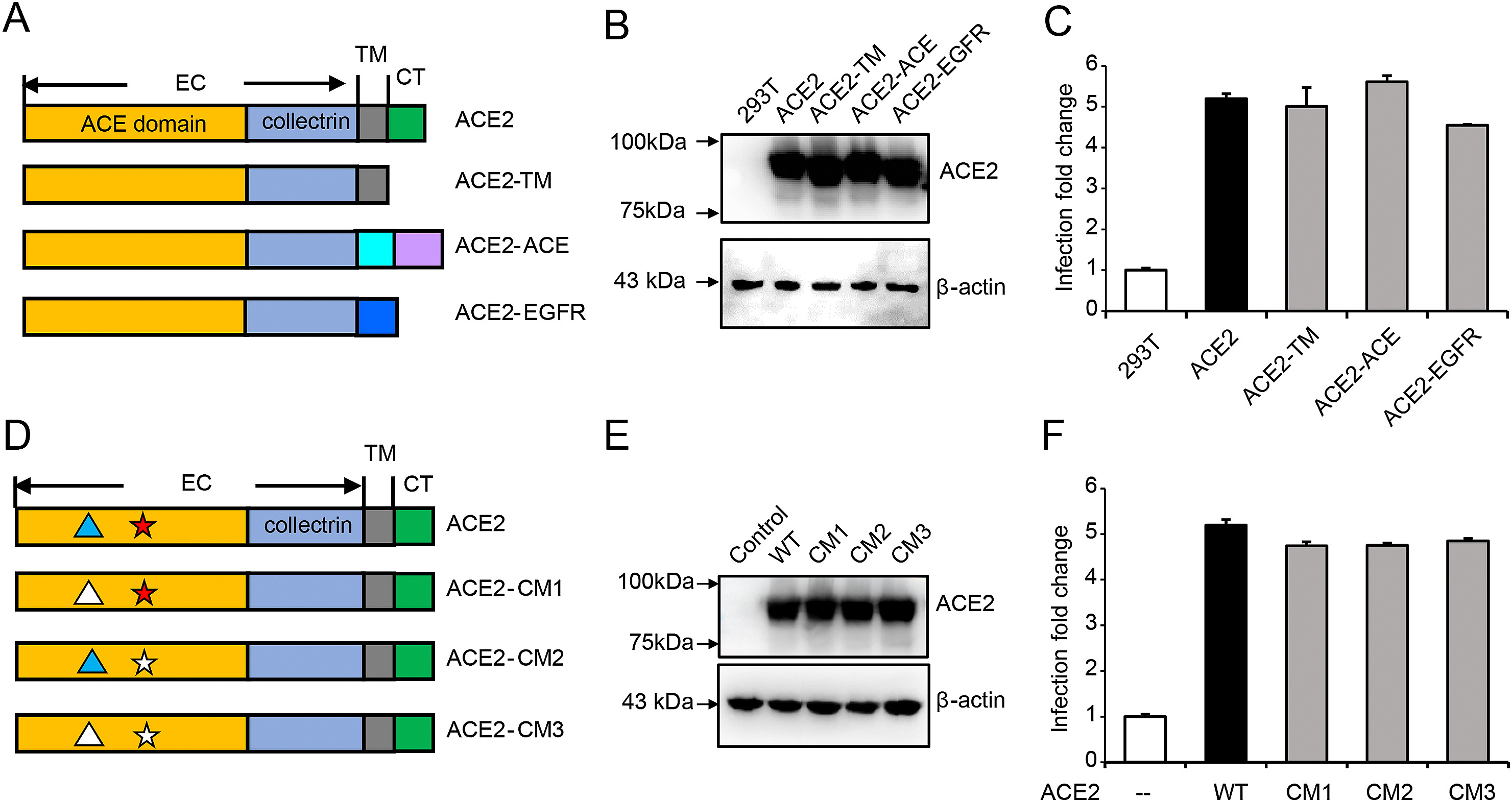
Hang Yang, Huijun Yuan, Xiaohui Zhao, Meng Xun, Shangrui Guo, Nan Wang, Bing Liu and Hongliang Wang. Cytoplasmic domain and enzymatic activity of ACE2 are not required for PI4KB dependent endocytosis entry of SARS-CoV-2 into host cells[J]. Virologica Sinica, 2022, 37(3): 380-389. doi: 10.1016/j.virs.2022.03.003.
The recent COVID-19 pandemic poses a global health emergency. Cellular entry of the causative agent SARS-CoV-2 is mediated by its spike protein interacting with cellular receptor-human angiotensin converting enzyme 2 (ACE2). Here, by using lentivirus based pseudotypes bearing spike protein, we demonstrated that entry of SARS-CoV-2 into host cells was dependent on clathrin-mediated endocytosis, and phosphoinositides played essential roles during this process. In addition, we showed that the intracellular domain and the catalytic activity of ACE2 were not required for efficient virus entry. Finally, we showed that the current predominant Delta variant, although with high infectivity and high syncytium formation, also entered cells through clathrin-mediated endocytosis. These results provide new insights into SARS-CoV-2 cellular entry and present proof of principle that targeting viral entry could be an effective way to treat different variant infections.
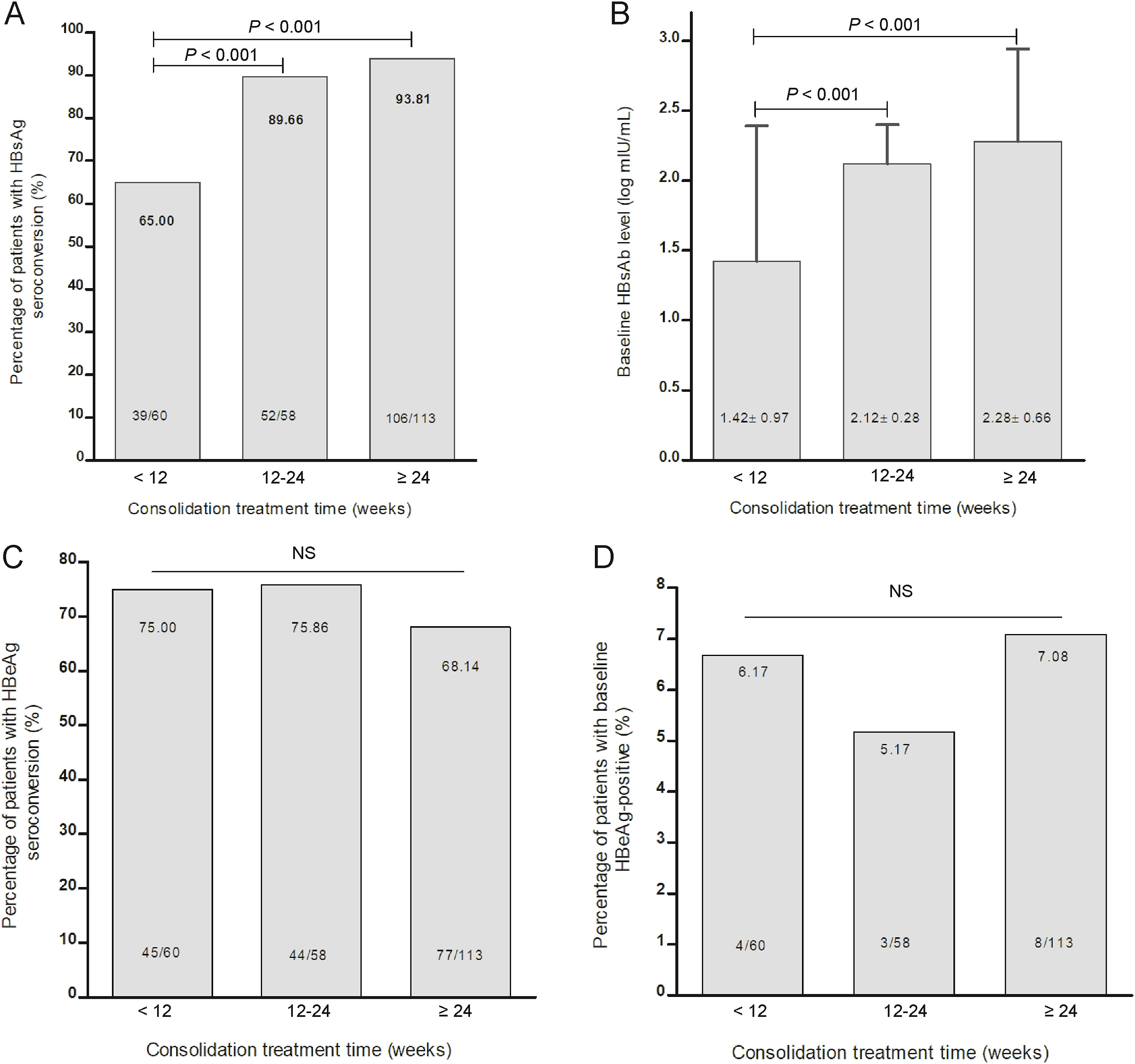
Minghui Li, Fangfang Sun, Xiaoyue Bi, Yanjie Lin, Liu Yang, Yao Lu, Lu Zhang, Gang Wan, Wei Yi, Linqing Zhao and Yao Xie. Consolidation treatment needed for sustained HBsAg-negative response induced by interferon-alpha in HBeAg positive chronic hepatitis B patients[J]. Virologica Sinica, 2022, 37(3): 390-397. doi: 10.1016/j.virs.2022.03.001.
Hepatitis B surface antigen (HBsAg) clearance is considered as functional cure in patients with chronic hepatitis B (CHB). This study aimed to assess the durability of HBsAg clearance achieved by interferon-based therapies in patients with CHB who were originally positive for hepatitis B envelope antigen (HBeAg). In this prospective study, HBeAg-positive CHB patients with confirmed HBsAg loss under interferon-based therapies were enrolled within 12 weeks from end of treatment and followed up for 48 weeks. Virological markers, biochemical indicators, and liver imaging examinations were observed every 3-6 months. Sustained functional cure was analysed as primary outcome. Factor associated with sustained HBsAg loss or reversion was also investigated. The rate of HBsAg loss sustainability was 91.8% (212/231). Patients receiving consolidation treatment for 12-24 weeks or ≥ 24 weeks had higher rates of sustained HBsAg negativity than those receiving consolidation treatment for < 12 weeks (98.3% and 91.2% vs. 86.7%, P=0.068), and the former groups had significantly higher anti-HBs levels than the later (P < 0.05). The cumulative incidence of HBsAg reversion and HBV DNA reversion was 8.2% and 3.9%, respectively. Consolidation treatment of ≥ 12 weeks[odd ratio (OR) 3.318, 95% confidence interval (CI) 1.077-10.224, P=0.037) was a predictor of sustained functional cure, and HBeAg-positivity at cessation of treatment (OR 12.271, 95% CI 1.076-139.919, P=0.043) was a predictor of HBsAg reversion. Interferon-alpha induced functional cure was durable and a consolidation treatment of ≥ 12-24 weeks was needed after HBsAg loss in HBeAg-positive CHB patients.
Tingting Liu, Anlei Liu, Yong Liu, Shan Cen and Quan Zhang. In vitro investigation of HBV clinical isolates from Chinese patients reveals that genotype C isolates possess higher infectivity than genotype B isolates[J]. Virologica Sinica, 2022, 37(3): 398-407. doi: 10.1016/j.virs.2022.03.008.
Hepatitis B virus (HBV) genotype B and C are two major genotypes that are prevalent in Asia and differ in natural history and disease progression. The impact of HBV genotypes on viral replication and protein expression has been explored by the transfection of hepatoma cells with replication-competent HBV DNA, which mimics the later stages of the viral life cycle. However, the influence of HBV genotypes on the early events of viral infection remains undetermined, mainly due to the difficulties in obtaining sufficient infectious viral particles for infection assays. Here, we report that a high-titer HBV inoculum can be generated from the transient transfection-based cell model after optimizing transfection conditions and modifying the HBV-expressing construct. By performing in vitro infection assays using transiently transfected derived viruses, we found that clinical genotype C isolates possessed higher infectivity than genotype B isolates. Moreover, we identified a naturally occurring mutation sL21S in small hepatitis B surface protein, which markedly decreased the infectivity of HBV genotype C isolates, but not that of genotype B isolates. In summary, using infectious viral particles provided by the optimized transient transfection-based cell model, we have been able to investigate a wide range of HBV variants on viral infectivity, which may contribute to our understanding of the reasons for different clinical outcomes in HBV infections and the development of therapeutic drugs targeting the early stages of HBV life cycle.
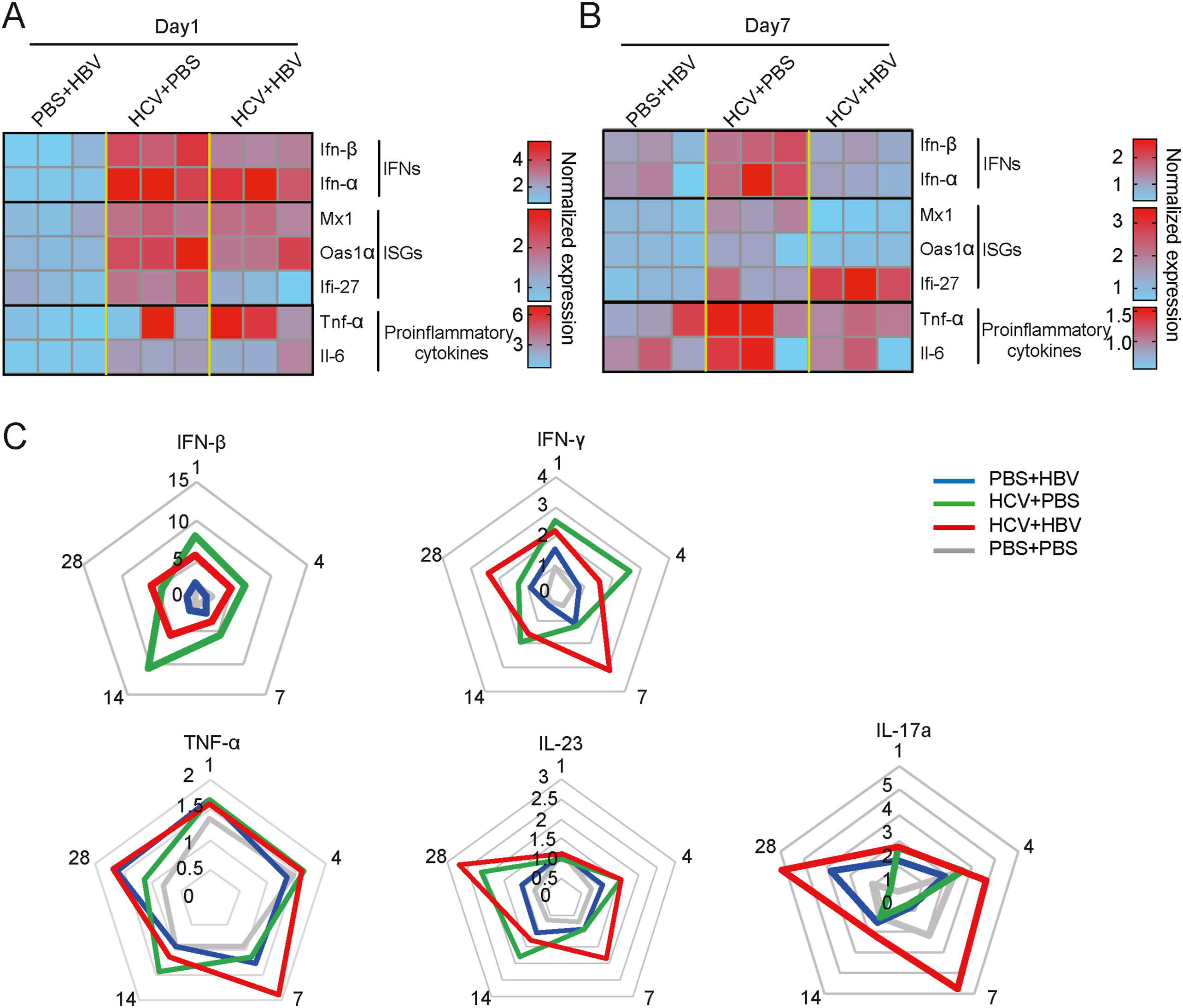
Shuhui Liu, Kaitao Zhao, Xi Su, Xiaoxiao Gao, Yongxuan Yao, Ranran Kong, Yun Wang, Chunchen Wu, Mengji Lu, Xinwen Chen and Rongjuan Pei. Enhanced host immune responses in presence of HCV facilitate HBV clearance in coinfection[J]. Virologica Sinica, 2022, 37(3): 408-417. doi: 10.1016/j.virs.2022.04.001.
Hepatitis B virus (HBV)/Hepatitis C virus (HCV) coinfection is frequently observed because of the common infection routine. Despite the reciprocal inhibition exerted by HBV and HCV genomes, the coinfection of HBV and HCV is associated with more severe forms of liver diseases. However, the complexity of viral interference and underlying pathological mechanism is still unclarified. With the demonstration of absence of direct viral interplay, some in vitro studies suggest the indirect effects of viral-host interaction on viral dominance outcome. Here, we comprehensively investigated the viral replication and host immune responses which might mediate the interference between viruses in HBV/HCV coinfected Huh7-NTCP cells and immunocompetent HCV human receptors transgenic ICR mice. We found that presence of HCV significantly inhibited HBV replication in vitro and in vivo irrespective of the coinfection order, while HBV did not affect HCV replication. Pathological alteration was coincidently reproduced in coinfected mice. In addition to the participation of innate immune response, an involvement of HCV in up-regulating HBV-specific immune responses was described to facilitate HBV clearance. Our systems partially recapitulate HBV/HCV coinfection and unveil the uncharacterized adaptive anti-viral immune responses during coinfection, which renews the knowledge on the nature of indirect viral interaction during HBV/HCV coinfection.
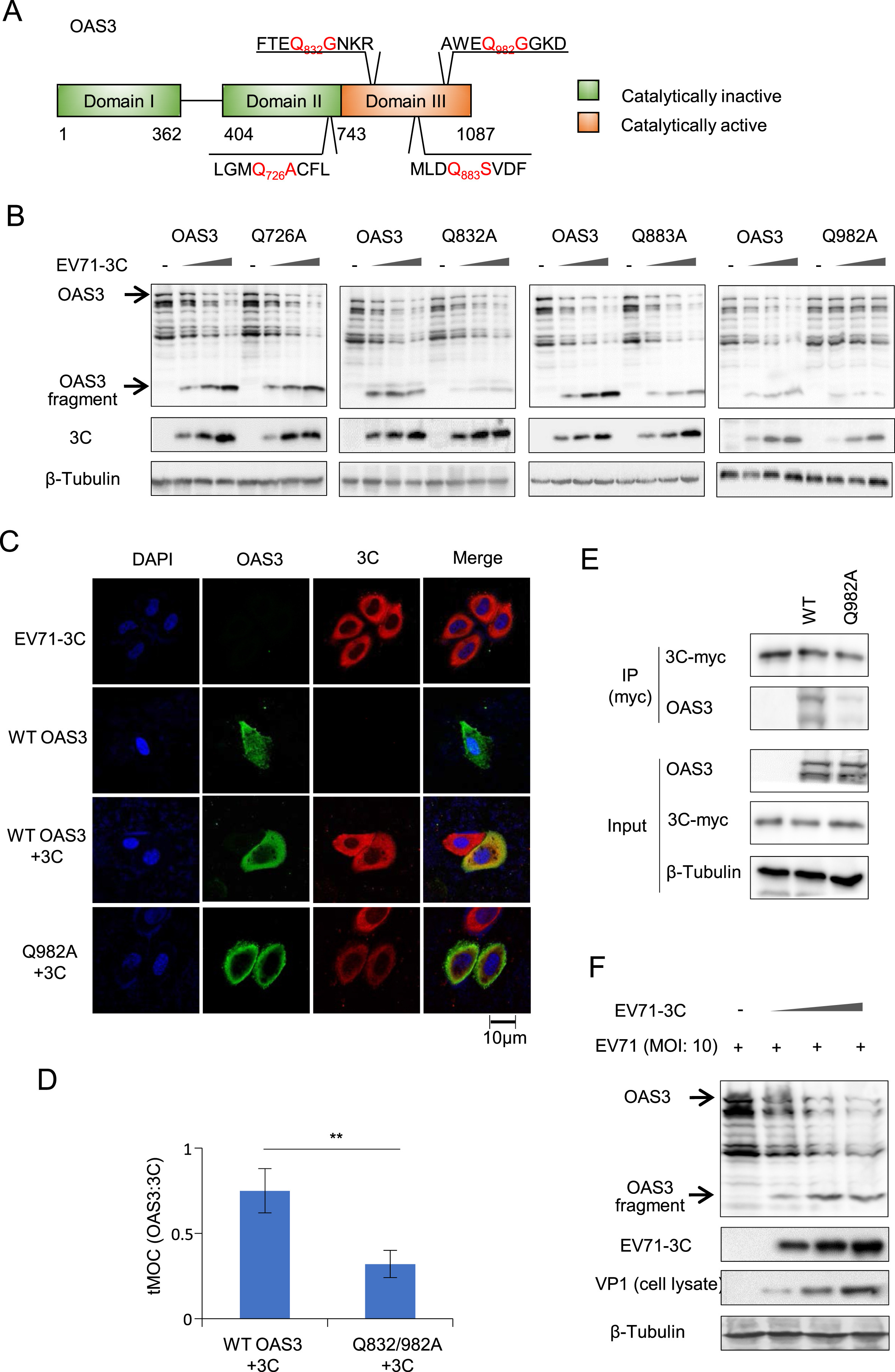
Xiaolei Zhou, Li Tian, Jian Wang, Baisong Zheng and Wenyan Zhang. EV71 3C protease cleaves host anti-viral factor OAS3 and enhances virus replication[J]. Virologica Sinica, 2022, 37(3): 418-426. doi: 10.1016/j.virs.2022.04.013.
The global spread of enteroviruses (EVs) has become more frequent, severe and life-threatening. Intereron (IFN) I has been proved to control EVs by regulating IFN-stimulated genes (ISG) expression. 20-50-oligoadenylate synthetases 3 (OAS3) is an important ISG in the OAS/RNase L antiviral system. The relationship between OAS3 and EVs is still unclear. Here, we reveal that OAS3, superior to OAS1 and OAS2, significantly inhibited EV71 replication in vitro. However, EV71 utilized autologous 3C protease (3Cpro) to cleave intracellular OAS3 and enhance viral replication. Rupintrivir, a human rhinovirus 3C protease inhibitor, completely abolished the cleavage of EV71 3Cpro on OAS3. And the proteolytically deficient mutants H40G, E71A, and C147G of EV71 3Cpro also lost the ability of OAS3 cleavage. Mechanistically, the Q982-G983 motif in C-terminal of OAS3 was identified as a crucial 3Cpro cutting site. Further investigation indicated that OAS3 inhibited not only EV71 but also Coxsackievirus B3 (CVB3), Coxsackievirus A16 (CA16), Enterovirus D68 (EVD68), and Coxsackievirus A6 (CA6) subtypes. Notably, unlike other four subtypes, CA16 3Cpro could not cleave OAS3. Two key amino acids variation Ile36 and Val86 in CA16 3Cpro might result in weak and delayed virus replication of CA16 because of failure of OAS and 3AB cleavage. Our works elucidate the broad anti-EVs function of OAS3, and illuminate a novel mechanism by which EV71 use 3Cpro to escape the antiviral effect of OAS3. These findings can be an important entry point for developing novel therapeutic strategies for multiple EVs infection.
Assane Hamidou Abdoulaye, Jichun Jia, Aqleem Abbas, Du Hai, Jiasen Cheng, Yanping Fu, Yang Lin, Daohong Jiang and Jiatao Xie. Fusarivirus accessory helicases present an evolutionary link for viruses infecting plants and fungi[J]. Virologica Sinica, 2022, 37(3): 427-436. doi: 10.1016/j.virs.2022.03.010.
A significant number of mycoviruses have been identified that are related to plant viruses, but their evolutionary relationships are largely unexplored. A fusarivirus, Rhizoctonia solani fusarivirus 4 (RsFV4), was identified in phytopathogenic fungus Rhizoctonia solani (R. solani) strain XY74 co-infected by an alphaendornavirus. RsFV4 had a genome of 10,833 nt (excluding the poly-A tail), and consisted of four non-overlapping open reading frames (ORFs). ORF1 encodes an 825 aa protein containing a conserved helicase domain (Hel1). ORF3 encodes 1550 aa protein with two conserved domains, namely an RNA-dependent RNA polymerase (RdRp) and another helicase (Hel2). The ORF2 and ORF4 likely encode two hypothetical proteins (520 and 542 aa) with unknown functions. The phylogenetic analysis based on Hel2 and RdRp suggest that RsFV4 was positioned within the fusarivirus group, but formed an independent branch with three previously reported fusariviruses of R. solani. Notably, the Hel1 and its relatives were phylogenetically closer to helicases of potyviruses and hypoviruses than fusariviruses, suggesting fusarivirus Hel1 formed an evolutionary link between these three virus groups. This finding provides evidence of the occurrence of a horizontal gene transfer or recombination event between mycoviruses and plant viruses or between mycoviruses. Our findings are likely to enhance the understanding of virus evolution and diversity.
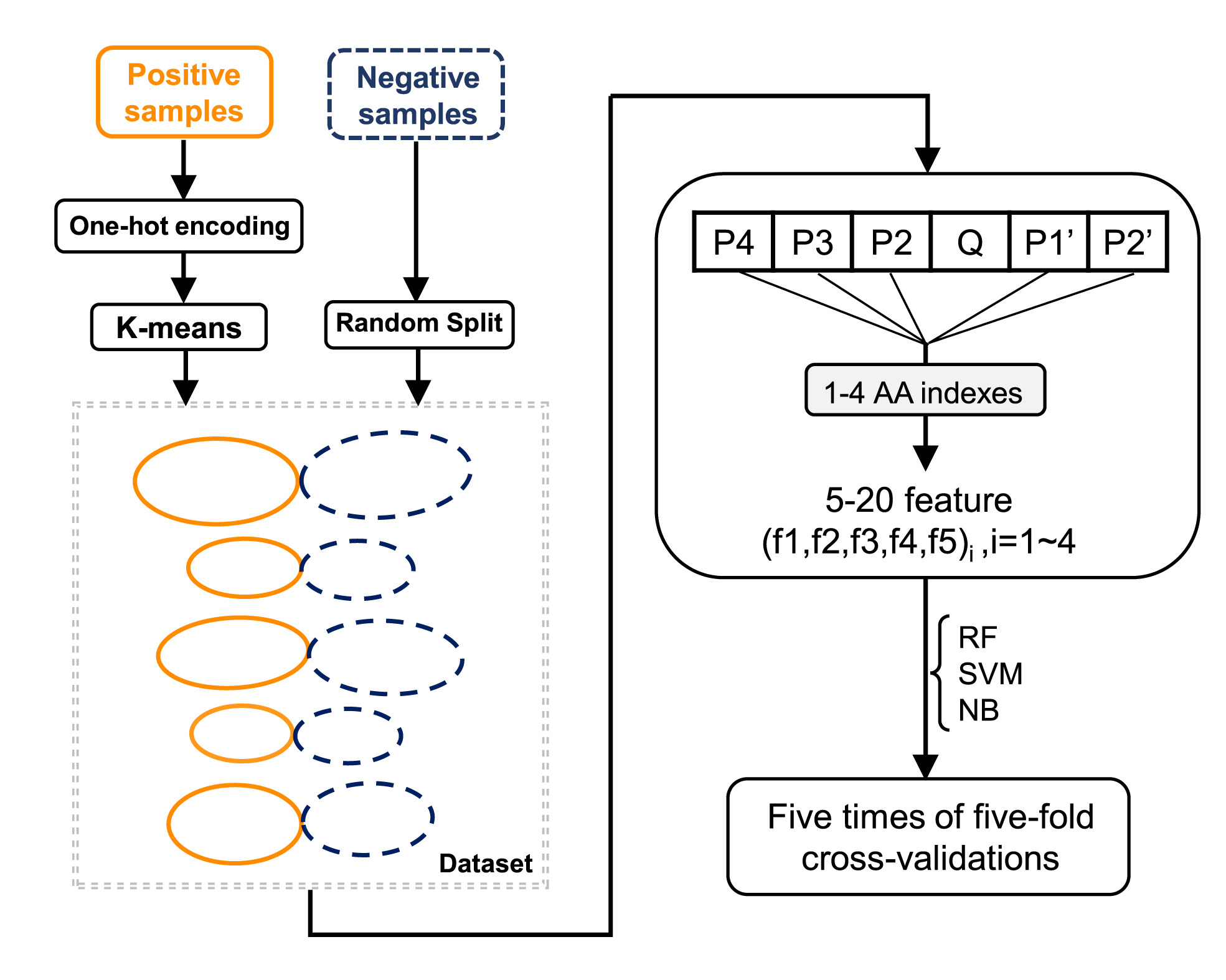
Huiting Chen, Zhaozhong Zhu, Ye Qiu, Xingyi Ge, Heping Zheng and Yousong Peng. Prediction of coronavirus 3C-like protease cleavage sites using machine-learning algorithms[J]. Virologica Sinica, 2022, 37(3): 437-444. doi: 10.1016/j.virs.2022.04.006.
The coronavirus 3C-like (3CL) protease, a cysteine protease, plays an important role in viral infection and immune escape. However, there is still a lack of effective tools for determining the cleavage sites of the 3CL protease. This study systematically investigated the diversity of the cleavage sites of the coronavirus 3CL protease on the viral polyprotein, and found that the cleavage motif were highly conserved for viruses in the genera of Alphacoronavirus, Betacoronavirus and Gammacoronavirus. Strong residue preferences were observed at the neighboring positions of the cleavage sites. A random forest (RF) model was built to predict the cleavage sites of the coronavirus 3CL protease based on the representation of residues in cleavage motifs by amino acid indexes, and the model achieved an AUC of 0.96 in cross-validations. The RF model was further tested on an independent test dataset which were composed of cleavage sites on 99 proteins from multiple coronavirus hosts. It achieved an AUC of 0.95 and predicted correctly 80% of the cleavage sites. Then, 1,352 human proteins were predicted to be cleaved by the 3CL protease by the RF model. These proteins were enriched in several GO terms related to the cytoskeleton, such as the microtubule, actin and tubulin. Finally, a webserver named 3CLP was built to predict the cleavage sites of the coronavirus 3CL protease based on the RF model. Overall, the study provides an effective tool for identifying cleavage sites of the 3CL protease and provides insights into the molecular mechanism underlying the pathogenicity of coronaviruses.
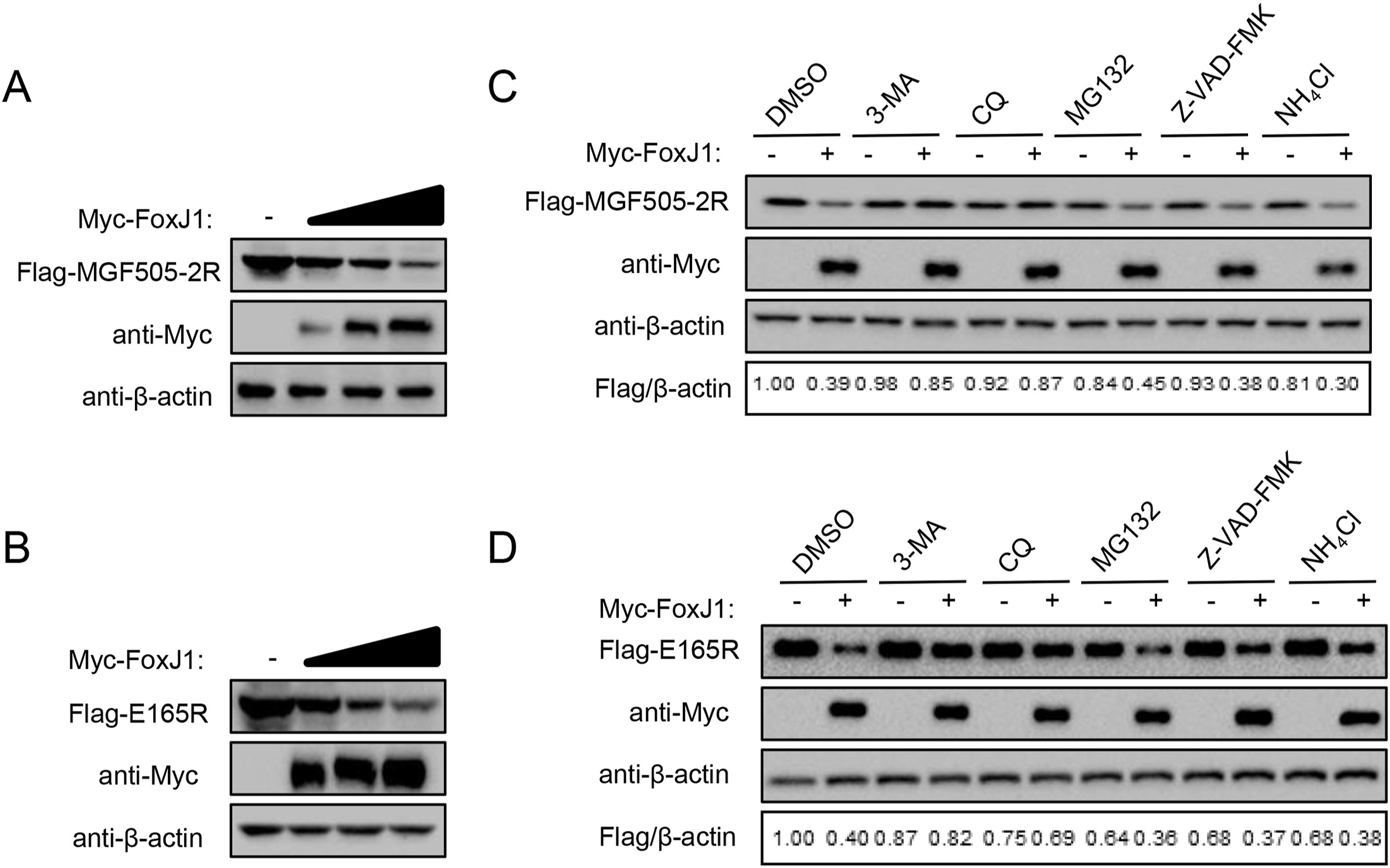
Caina Ma, Shasha Li, Fan Yang, Weijun Cao, Huisheng Liu, Tao Feng, Keshan Zhang, Zixiang Zhu, Xiangtao Liu, Yonghao Hu and Haixue Zheng. FoxJ1 inhibits African swine fever virus replication and viral S273R protein decreases the expression of FoxJ1 to impair its antiviral effect[J]. Virologica Sinica, 2022, 37(3): 445-454. doi: 10.1016/j.virs.2022.04.008.
African swine fever (ASF) is a highly pathogenic swine infectious disease that affects domestic pigs and wild boar, which is caused by the African swine fever virus (ASFV). ASF has caused huge economic losses to the pig industry and seriously threatens global food security and livestock health. To date, there is no safe and effective commercial vaccine against ASF. Unveiling the underlying mechanisms of ASFV-host interplay is critical for developing effective vaccines and drugs against ASFV. In the present study, RNA-sequencing, RT-qPCR and Western blotting analysis revealed that the transcriptional and protein levels of the host factor FoxJ1 were significantly down-regulated in primary porcine alveolar macrophages (PAMs) infected by ASFV. RT-qPCR analysis showed that overexpression of FoxJ1 upregulated the transcription of type I interferon and interferon stimulating genes (ISGs) induced by poly(dA:dT). FoxJ1 revealed a function to positively regulate innate immune response, therefore, suppressing the replication of ASFV. In addition, Western blotting analysis indicated that FoxJ1 degraded ASFV MGF505-2R and E165R proteins through autophagy pathway. Meanwhile, RT-qPCR and Western blotting analysis showed that ASFV S273R inhibited the expression of FoxJ1. Altogether, we determined that FoxJ1 plays an antiviral role against ASFV replication, and ASFV protein impairs FoxJ1-mediated antiviral effect by degradation of FoxJ1. Our findings provide new insights into the antiviral function of FoxJ1, which might help design antiviral drugs or vaccines against ASFV infection.
Minglong Liang, Zongmei Wang, Chuanjian Wu, Sidong Xiong, Ling Zhao and Chunsheng Dong. A single dose of recombinant VSV-RABVG vaccine provides full protection against RABV challenge[J]. Virologica Sinica, 2022, 37(3): 455-458. doi: 10.1016/j.virs.2022.02.008.
Highlights: 1. A replication-competent recombinant VSV with RABV-G protein replacement was generated. 2. Single dose of VSV-RABVG immunization induce potent antigen-specific humoral immune response, especially the virus neutralizing antibodies. 3. Mice intranasally immunized with single dose of VSV-RABVG were 100% protected upon RABV challenge.
Yuan Zhang, Chunjie Li, Xianliang Ke, Dan Luo, Yan Liu, Quanjiao Chen, Hanzhong Wang, Xiaohui Song and Zhenhua Zheng. Development of a biosensor assessing SARS-CoV-2 main protease proteolytic activity in living cells for antiviral drugs screening[J]. Virologica Sinica, 2022, 37(3): 459-461. doi: 10.1016/j.virs.2022.04.002.
Highlights: The biosensor reported in our study can monitor SARS-CoV-2 Mpro activity in living cells instead of in vitro solutions. The biosensor reported in our study is sensitive and easy to operate. It is suitable for high-throughput screening. It has the potential to be used in small animal models.
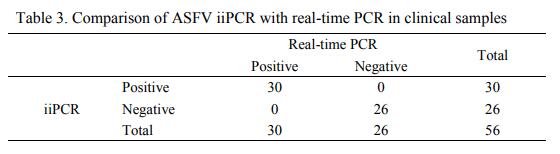
Tianli Zou, Junhua Deng, Xiangdong Li, Shiyin Zhang, Lingyan Chen, Liying Hao, Jinshan Zhuang, Heng Wang, Guihong Zhang, Shengxiang Ge and Kegong Tian. Development of a fluorescent probe hydrolysis-insulated isothermal PCR for rapid and sensitive on-site detection of African swine fever virus[J]. Virologica Sinica, 2022, 37(3): 462-464. doi: 10.1016/j.virs.2022.03.002.
Highlights 1. A probe-based insulated isothermal PCR (iiPCR) assay was developed for rapid and onsite detection of ASFV. 2. The developed iiPCR showed similar sensitivity and specificity with OIE recommended real-time PCR. 3. Blood samples could be directly applied as PCR template in iiPCR without DNA extraction.
Highlights · Highthrouput sequencing of small RNA of H5N1 infected and mock infected chicken lungs. · 297 miRNAs identified in mock-infected and 201 miRNAs identified in AIV infected chicken lungs. · 36 miRNAs were upregulated and 90 were downregulated during H5N1 infection. · Functional analysis and gene ontology of predicted target genes of expressed miRNAs. · MAPK pathway, NF-κB, IGF and gga-let-7b might play important role during H5N1 pathogenesis.
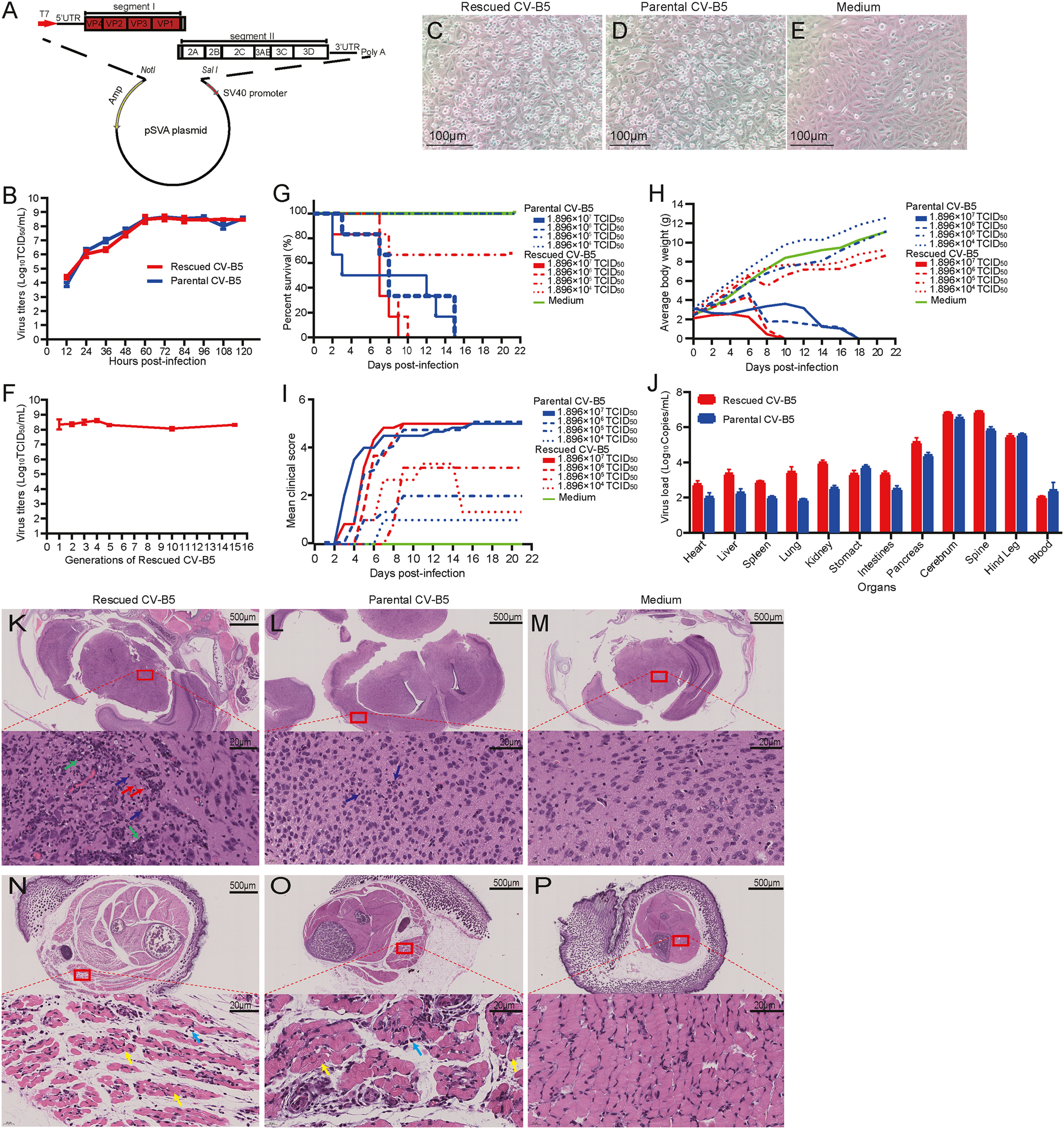
Lifang Song, Bopei Cui, Jinghuan Yang, Xiaotian Hao, Xujia Yan, Jialu Zhang, Dong Liu, Ziyang Song, Qian Wang, Qunying Mao and Zhenglun Liang. Construction and verification of an infectious cDNA clone of coxsackievirus B5[J]. Virologica Sinica, 2022, 37(3): 469-471. doi: 10.1016/j.virs.2022.03.005.
Highlights: · An infectious cDNA clone of CV-B5 was constructed. · The rescued and parental virus possessed similar biological characteristics. · The virulence of the rescued virus was similiar to that of the parental virus. · Viral distribution and tissue tropism of those two viruses were in agreement.
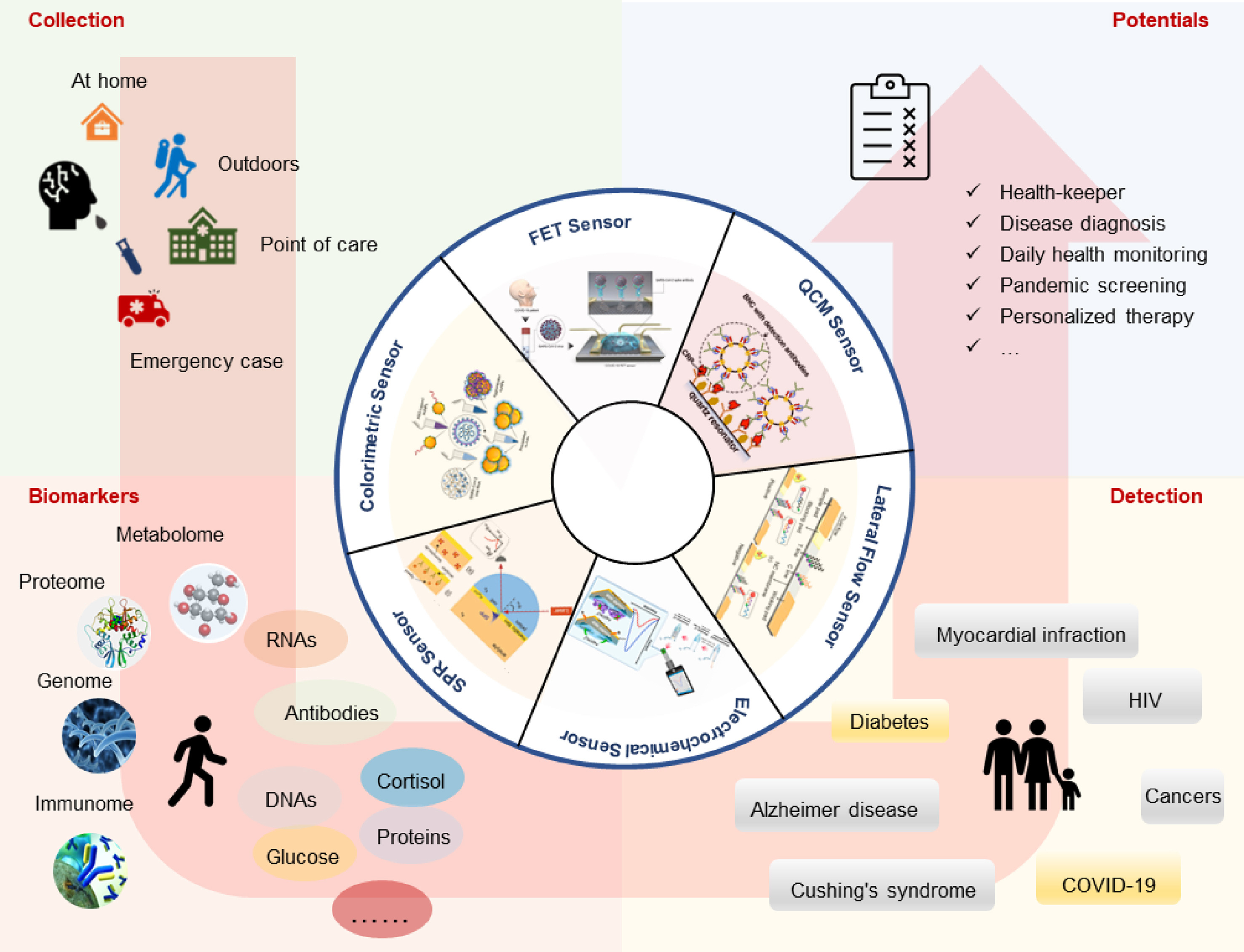
Shiwen Wang, Ying Liu, Yang Qiu, Qian Dou, Yang Han, Muhan Huang, Ke Hong, Bei Yang, Xi Zhou and Qing Dai. Saliva-based point-of-care testing techniques for COVID-19 detection[J]. Virologica Sinica, 2022, 37(3): 472-476. doi: 10.1016/j.virs.2022.04.004.
Highlights 1. The advantages of COVID-19 detection in saliva were systematically introduced. 2. Saliva-based POCT technologies for the detection of COVID-19 were reviewed. 3. A positive correlation between COVID-19 antibodies in saliva and serum was demonstrated.