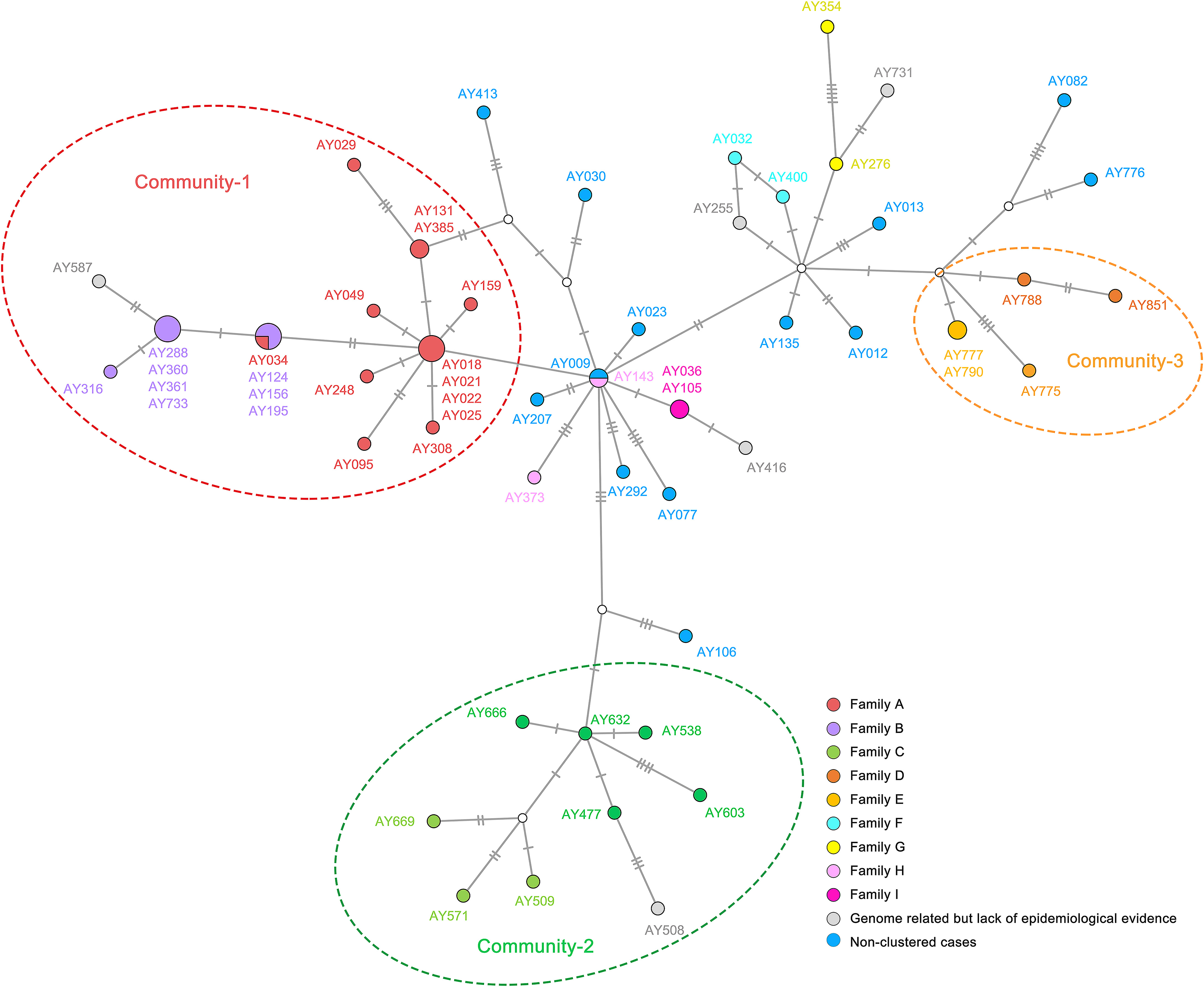
In the past two years, numerous newly emerging and threatening SARS-CoV-2 lineages have been identified in various countries and regions. Reconstructing transmission chains/networks has become an effective way to elucidate the dynamics and evolution of SARS-CoV-2. In this issue, Li et al. carried out a comprehensive analysis on the family and community SARS-CoV-2 transmission events in a representative small and medium-sized city, employing a combination of epidemiological investigation, high-throughput sequencing, and clinical testing. Their study describes a complete dynamic process of single-nucleotide polymorphism development during the community transmission, and further reveals that intrahost variant analysis is an effective approach to studying cluster infections. The cover image shows haplotype network based on SARS-CoV-2 genomes from different cases and transmission chains relied on epidemiological information (reconstructing transmission process works like revolving jigsaw puzzle; provided by Yang Li and Peng Zhou, designed by Yuxian Liu). Please see page 187–197 for details.
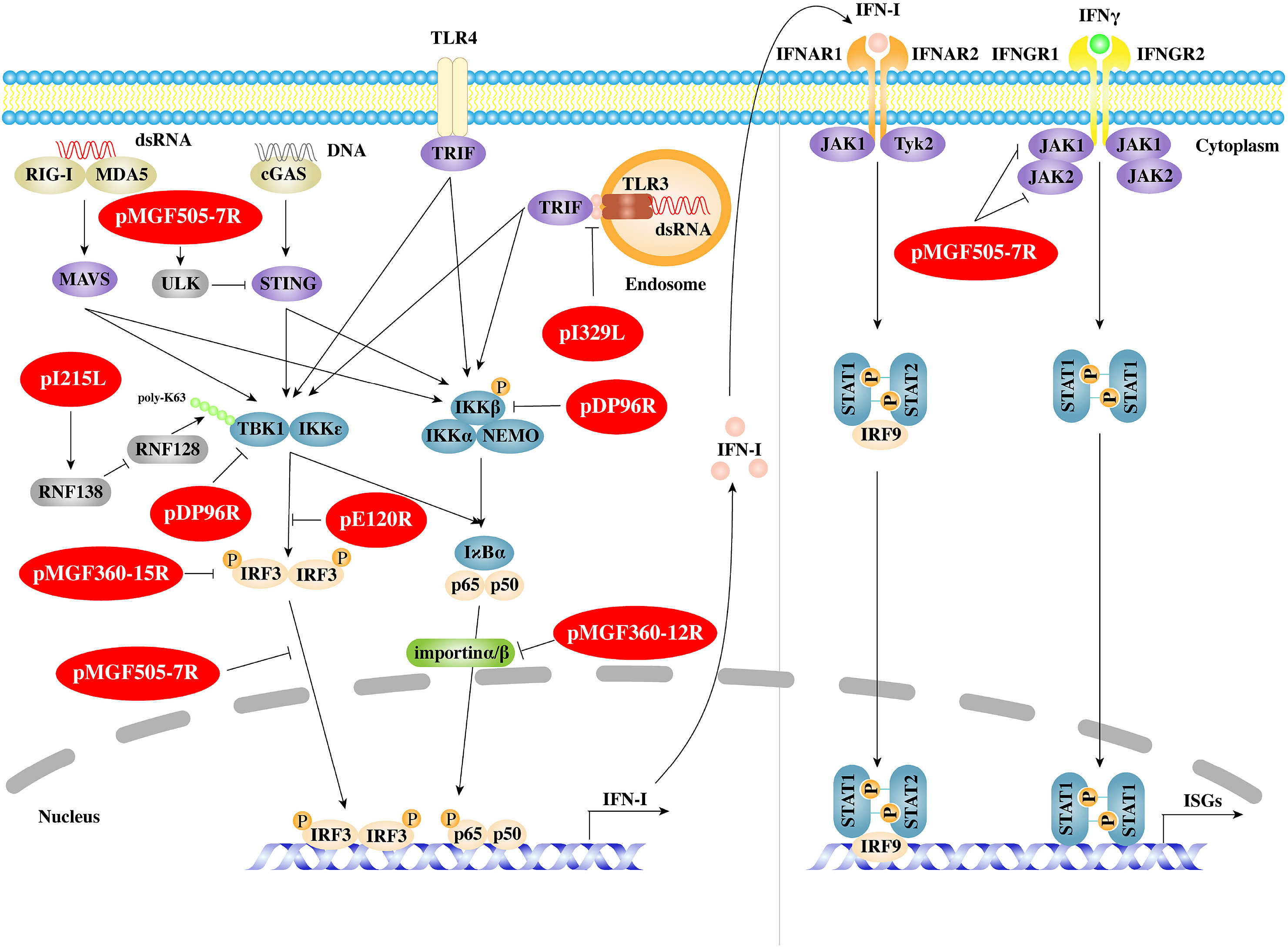
Xiaojie Zheng, Shengming Nie and Wen-Hai Feng. Regulation of antiviral immune response by African swine fever virus (ASFV)[J]. Virologica Sinica, 2022, 37(2): 157-167. doi: 10.1016/j.virs.2022.03.006.
African swine fever (ASF) is a highly contagious and acute hemorrhagic viral disease with a high mortality approaching 100% in domestic pigs. ASF is an endemic in countries in sub-Saharan Africa. Now, it has been spreading to many countries, especially in Asia and Europe. Due to the fact that there is no commercial vaccine available for ASF to provide sustainable prevention, the disease has spread rapidly worldwide and caused great economic losses in swine industry. The knowledge gap of ASF virus (ASFV) pathogenesis and immune evasion is the main factor to limit the development of safe and effective ASF vaccines. Here, we will summarize the molecular mechanisms of how ASFV interferes with the host innate and adaptive immune responses. An in-depth understanding of ASFV immune evasion strategies will provide us with rational design of ASF vaccines.
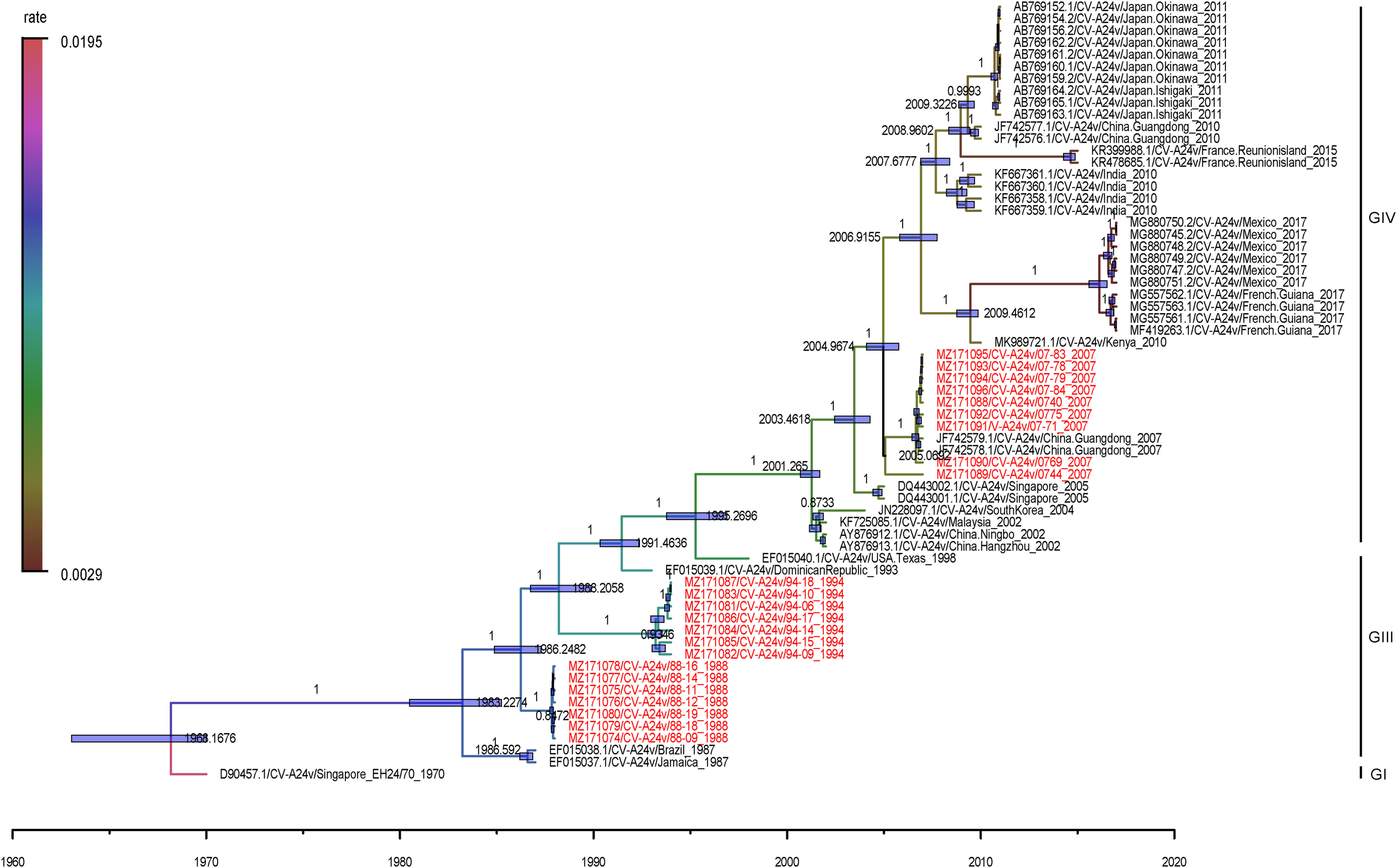
Junhan Li, Fang Huang, Yong Zhang, Tianjiao Ji, Shuangli Zhu, Dongyan Wang, Zhenzhi Han, Jinbo Xiao, Fenfen Si, Wenbo Xu and Dongmei Yan. Molecular analysis of Coxsackievirus A24 variant isolates from three outbreaks of acute hemorrhagic conjunctivitis in 1988, 1994 and 2007 in Beijing, China[J]. Virologica Sinica, 2022, 37(2): 168-176. doi: 10.1016/j.virs.2022.01.024.
Coxsackievirus A24 variant (CVA24v) is a major pathogen that causes continued outbreaks and pandemics of acute hemorrhagic conjunctivitis (AHC). In China, the first confirmed outbreak of CVA24v-related AHC occurred in Beijing in 1988, followed by another two significant outbreaks respectively in 1994 and 2007, which coincides with the three-stage dynamic distribution of AHC in the world after 1970s. To illustrate the genetic characteristics of CVA24v in different periods, a total of 23 strains were isolated from those three outbreaks and the whole genome of those isolations were sequenced and analyzed. Compared with the prototype strain, the 23 strains shared four nucleotide deletions in the 5' UTR except the 0744 strain isolated in 2007. And at the 98th site, one nucleotide insertion was found in all the strains collected from 2007. From 1994 to 2007, amino acid polarity in the VP1 region at the 25th and the 32nd site were changed. Both the 3C and VP1 phylogenetic tree indicated that isolates from 1988 and 1994 belonged to Genotype III (GIII), and 2007 strains to Genotype IV (GIV). According to the Bayesian analysis based on complete genome sequence, the most recent common ancestors for the isolates in 1988, 1994 and 2007 were respectively estimated around October 1987, February 1993 and December 2004. The evolutionary rate of the CVA24v was estimated to be 7.45×10-3 substitutions/site/year. Our study indicated that the early epidemic of CVA24v in Chinese mainland was the GIII. Point mutations and amino acid changes in different genotypes of CVA24v may generate intensity differences of the AHC outbreak. CVA24v has been evolving constantly with a relatively rapid rate.
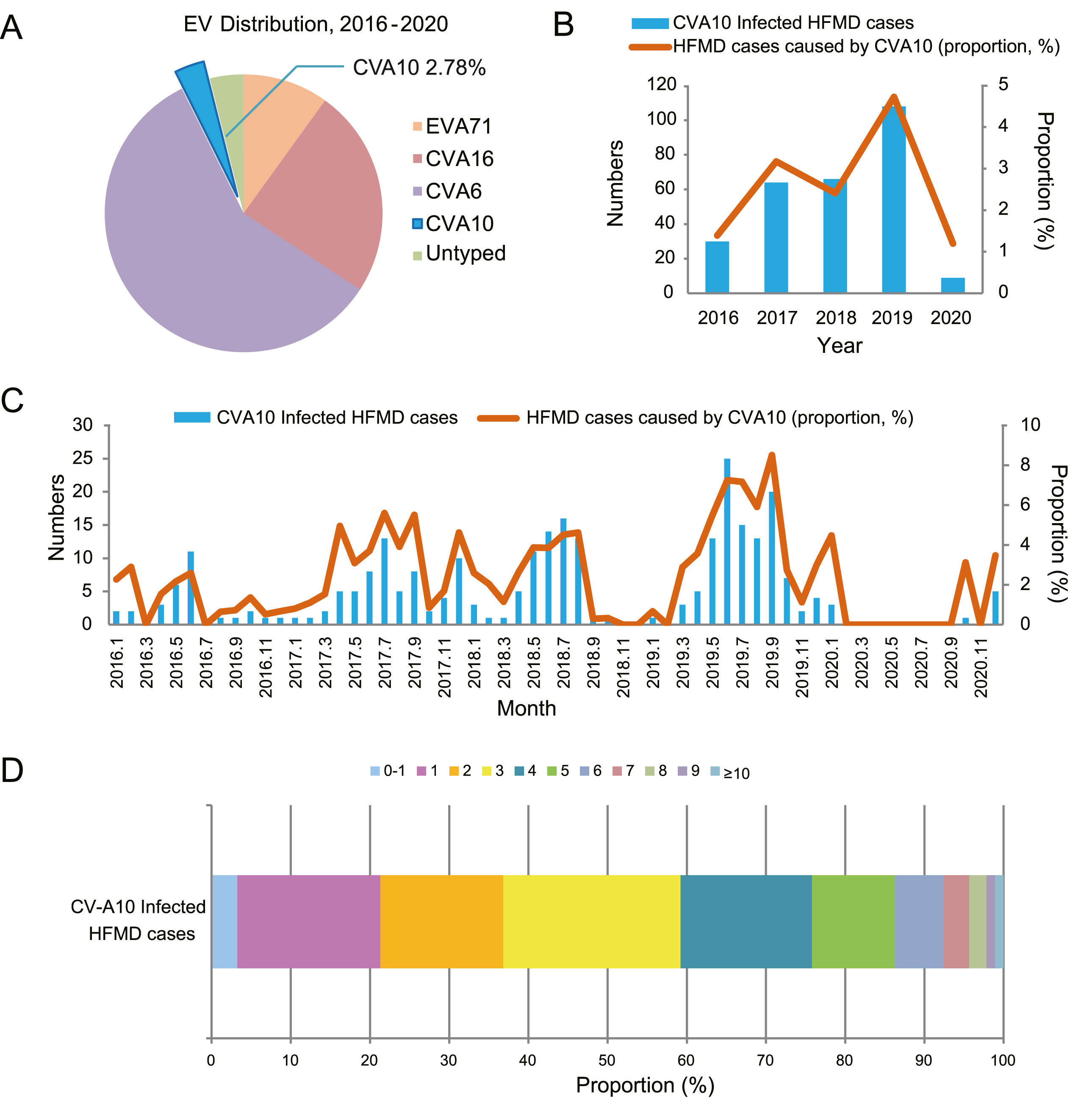
Jiayu Wang, Jiajing Liu, Fanghao Fang, Jiajin Wu, Tianjiao Ji, Yuying Yang, Ling Liu, Chongshan Li, Wanju Zhang, Xi Zhang and Zheng Teng. Genomic surveillance of coxsackievirus A10 reveals genetic features and recent appearance of genogroup D in Shanghai, China, 2016-2020[J]. Virologica Sinica, 2022, 37(2): 177-186. doi: 10.1016/j.virs.2022.01.028.
Coxsackievirus A10 (CVA10) is one of the major causative agents of hand, foot and mouth disease (HFMD). To investigate the epidemiological characteristics as well as genetic features of CVA10 currently circulating in Shanghai, China, we collected a total of 9,952 sporadic HFMD cases from January 2016 to December 2020. In the past five years, CVA10 was the fourth prevalent causatives associated with HFMD in Shanghai and the overall positive rate was 2.78%. The annual distribution experienced significant fluctuations over the past five years. In addition to entire VP1 sequencing, complete genome sequencing and recombination analysis of CVA10 isolates in Shanghai were further performed. A total of 64 near complete genomes and 11 entire VP1 sequences in this study combined with reference sequences publicly available were integrated into phylogenetic analysis. The CVA10 sequences in this study mainly belonged to genogroup C and presented 91%-100% nucleotide identity with other Chinese isolates based on VP1 region. For the first time, our study reported the appearance of CVA10 genogroup D in Chinese mainland, which had led to large-scale outbreaks in Europe previously. The recombination analysis showed the recombination break point located between 5,100 nt and 6,700 nt, which suggesting intertypic recombination with CVA16 genogroup D. To conclusion, CVA10 genogroup C was the predominant genogroup in Shanghai during 2016-2020. CVA10 recombinant genogroup D was firstly reported in circulating in Chinese mainland. Continuous surveillance is needed to better understand the evolution relationships and transmission pathways of CVA10 to help to guide disease control and prevention.
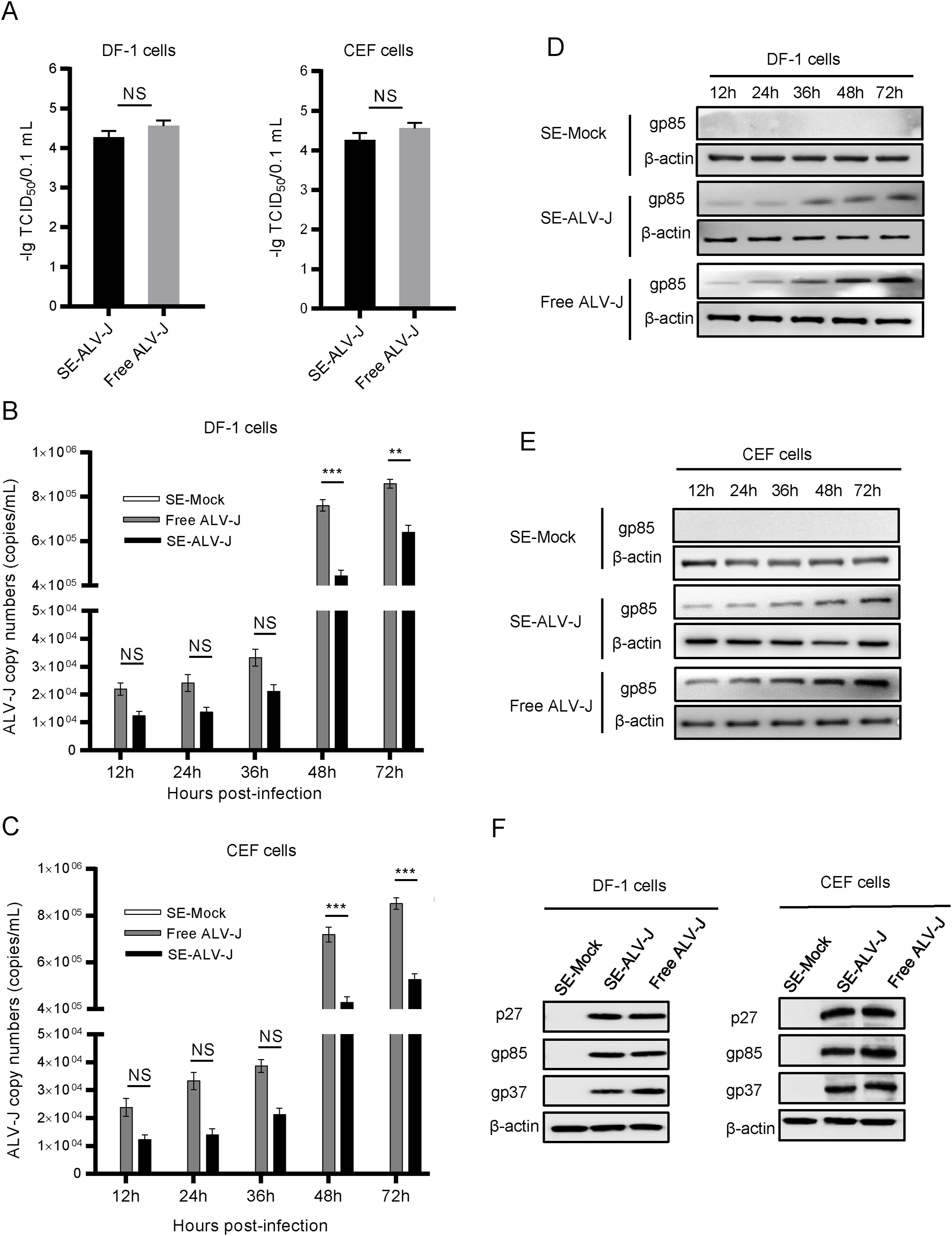
Yang Li, Hao-Rui Si, Yan Zhu, Nan Xie, Bei Li, Xiang-Ping Zhang, Jun-Feng Han, Hong-Hong Bao, Yong Yang, Kai Zhao, Zi-Yuan Hou, Si-Jia Cheng, Shuan-Hu Zhang, Zheng-Li Shi and Peng Zhou. Characteristics of SARS-CoV-2 transmission in a medium-sized city with traditional communities during the early COVID-19 epidemic in China[J]. Virologica Sinica, 2022, 37(2): 187-197. doi: 10.1016/j.virs.2022.01.030.
The nationwide COVID-19 epidemic ended in 2020, a few months after its outbreak in Wuhan, China at the end of 2019. Most COVID-19 cases occurred in Hubei Province, with a few local outbreaks in other provinces of China. A few studies have reported the early SARS-CoV-2 epidemics in several large cities or provinces of China. However, information regarding the early epidemics in small and medium-sized cities, where there are still traditionally large families and community culture is more strongly maintained and thus, transmission profiles may differ, is limited. In this study, we characterized 60 newly sequenced SARS-CoV-2 genomes from Anyang as a representative of small and medium-sized Chinese cities, compared them with more than 400 reference genomes from the early outbreak, and studied the SARS-CoV-2 transmission profiles. Genomic epidemiology revealed multiple SARS-CoV-2 introductions in Anyang and a large-scale expansion of the epidemic because of the large family size. Moreover, our study revealed two transmission patterns in a single outbreak, which were attributed to different social activities. We observed the complete dynamic process of single-nucleotide polymorphism development during community transmission and found that intrahost variant analysis was an effective approach to studying cluster infections. In summary, our study provided new SARS-CoV-2 transmission profiles representative of small and medium-sized Chinese cities as well as information on the evolution of SARS-CoV-2 strains during the early COVID-19 epidemic in China.
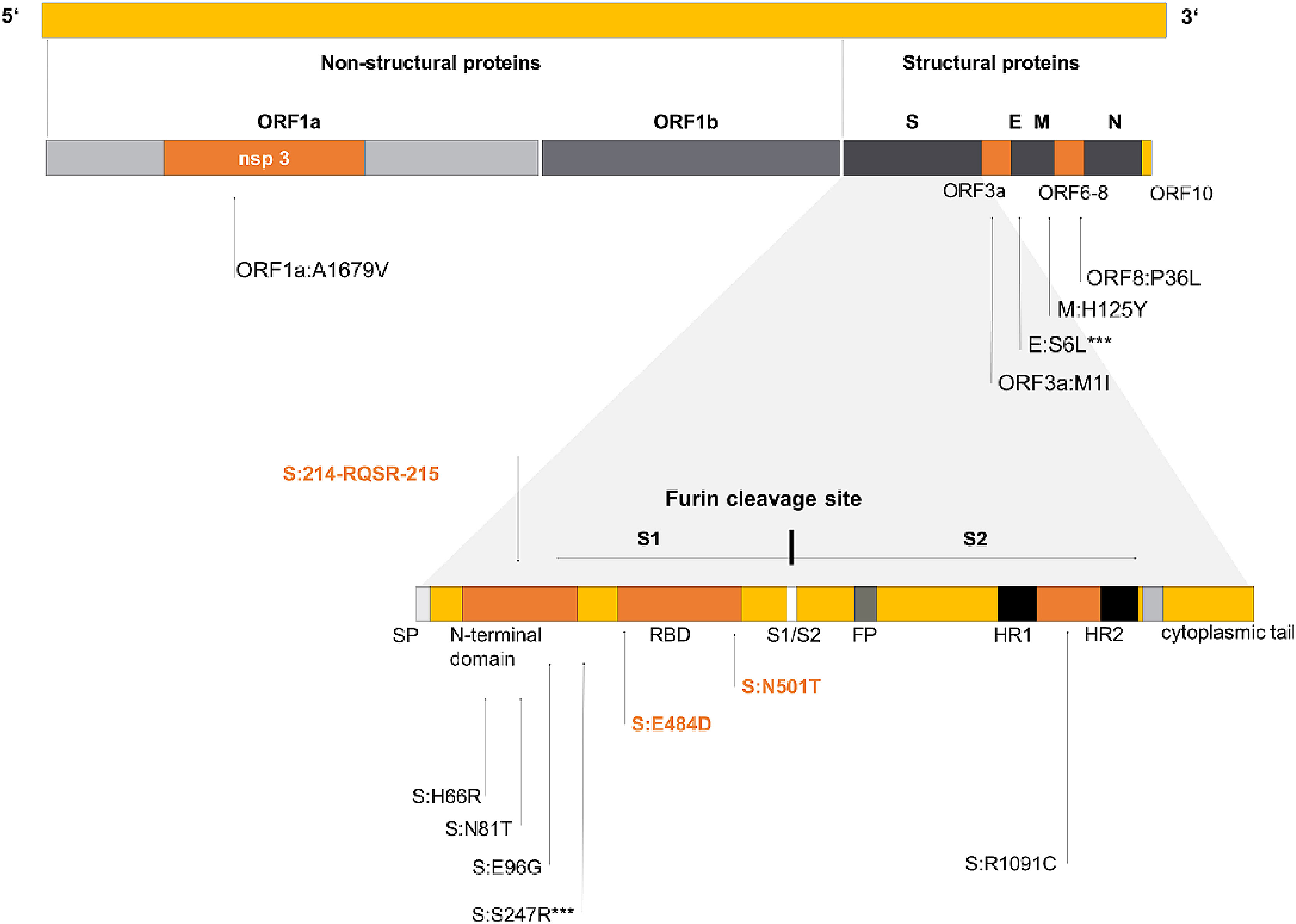
Sissy Therese Sonnleitner, Stefanie Sonnleitner, Eva Hinterbichler, Hannah Halbfurter, Dominik B.C. Kopecky, Stephan Koblmüller, Christian Sturmbauer, Wilfried Posch and Gernot Walder. The mutational dynamics of the SARS-CoV-2 virus in serial passages in vitro[J]. Virologica Sinica, 2022, 37(2): 198-207. doi: 10.1016/j.virs.2022.01.029.
Since its outbreak in 2019, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) keeps surprising the medical community by evolving diverse immune escape mutations in a rapid and effective manner. To gain deeper insight into mutation frequency and dynamics, we isolated ten ancestral strains of SARS-CoV-2 and performed consecutive serial incubation in ten replications in a suitable and common cell line and subsequently analysed them using RT-qPCR and whole genome sequencing. Along those lines we hoped to gain fundamental insights into the evolutionary capacity of SARS-CoV-2 in vitro. Our results identified a series of adaptive genetic changes, ranging from unique convergent substitutional mutations and hitherto undescribed insertions. The region coding for spike proved to be a mutational hotspot, evolving a number of mutational changes including the already known substitutions at positions S:484 and S:501. We discussed the evolution of all specific adaptations as well as possible reasons for the seemingly inhomogeneous potential of SARS-CoV-2 in the adaptation to cell culture. The combination of serial passage in vitro with whole genome sequencing uncovers the immense mutational potential of some SARS-CoV-2 strains. The observed genetic changes of SARS-CoV-2 in vitro could not be explained solely by selectively neutral mutations but possibly resulted from the action of directional selection accumulating favourable genetic changes in the evolving variants, along the path of increasing potency of the strain. Competition among a high number of quasi-species in the SARS-CoV-2 in vitro population gene pool may reinforce directional selection and boost the speed of evolutionary change.
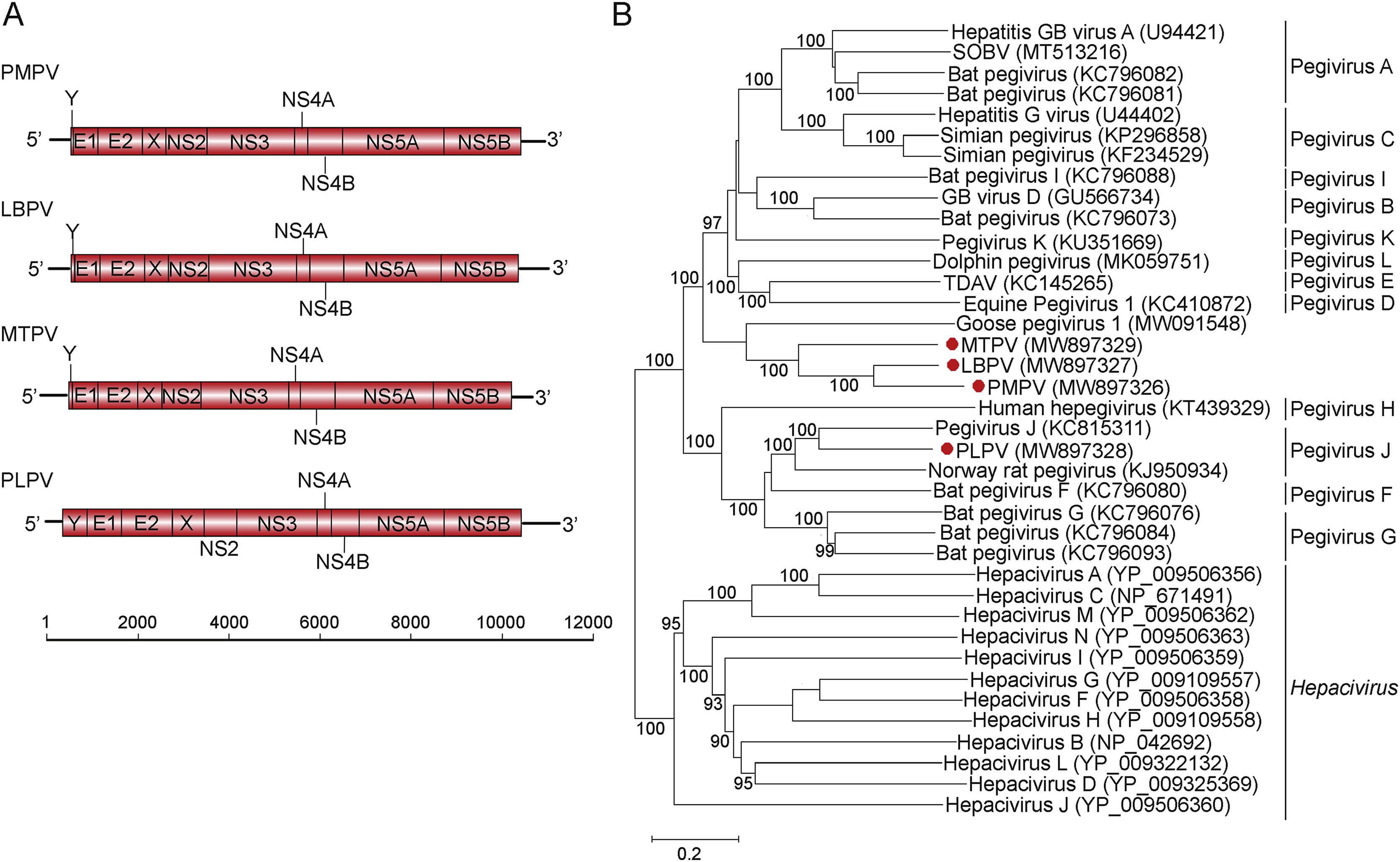
Wentao Zhu, Jing Yang, Shan Lu, Yuyuan Huang, Dong Jin, Ji Pu, Liyun Liu, Zhenjun Li, Mang Shi and Jianguo Xu. Novel pegiviruses infecting wild birds and rodents[J]. Virologica Sinica, 2022, 37(2): 208-214. doi: 10.1016/j.virs.2022.01.013.
Pegivirus (family Flaviviridae) is a genus of small enveloped RNA viruses that mainly causes blood infections in various mammals including human. Herein, we carried out an extensive survey of pegiviruses from a wide range of wild animals mainly sampled in the Qinghai-Tibet Plateau of China. Three novel pegiviruses, namely Passer montanus pegivirus, Leucosticte brandti pegivirus and Montifringilla taczanowskii pegivirus, were identified from different wild birds, and one new rodent pegivirus, namely Phaiomys leucurus pegivirus, was identified from Blyth's vole. Interestingly, the pegiviruses of non-mammalian origin discovered in this study substantially broaden the host range of Pegivirus to avian species. Co-evolutionary analysis showed virus-host co-divergence over long evolutionary timescales, and indicated that pegiviruses largely followed a virus-host co-divergence relationship. Overall, this work extends the biodiversity of the Pegivirus genus to those infecting wild birds and hence revises the host range and evolutionary history of genus Pegivirus.
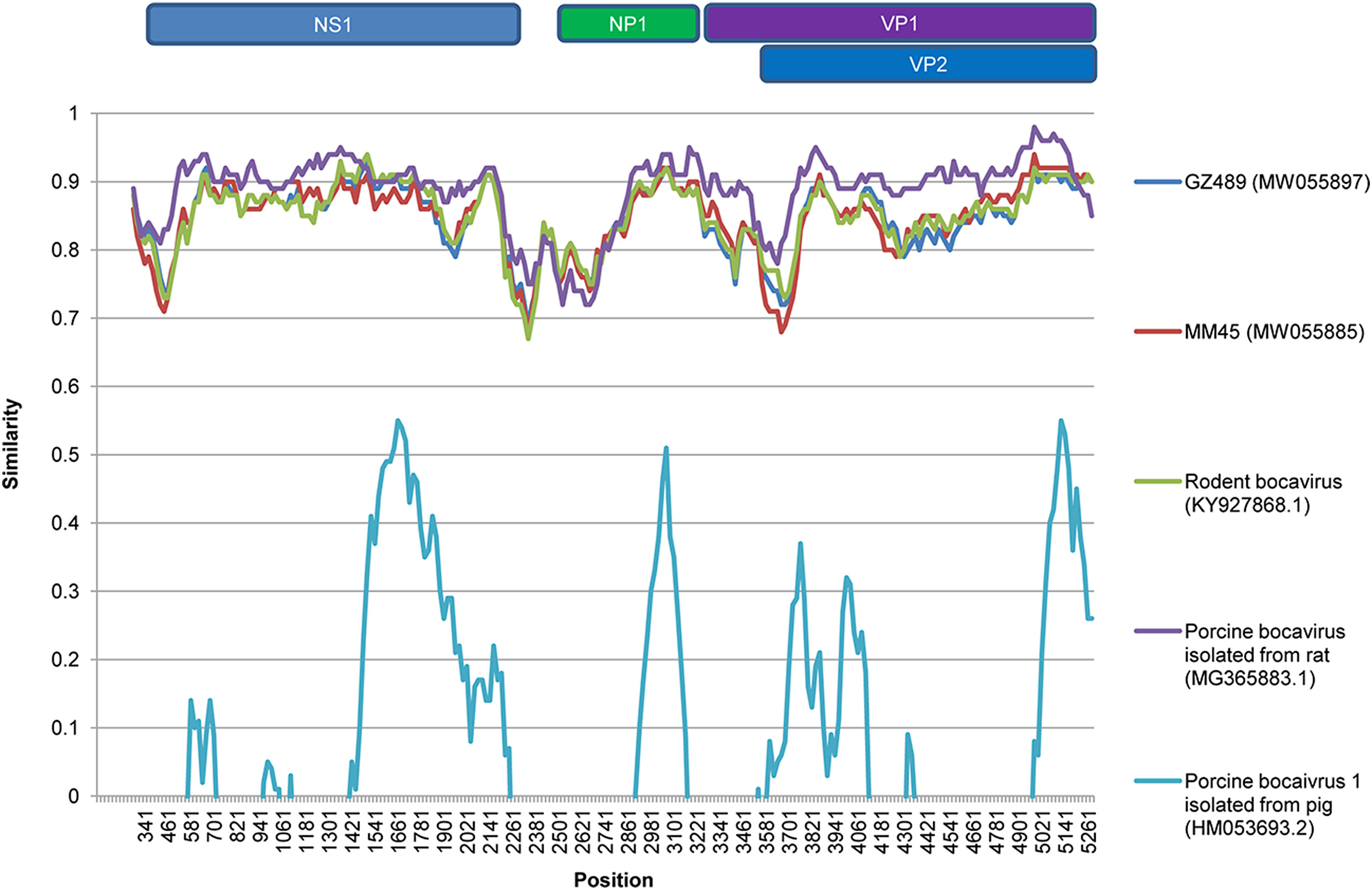
Wenqiao He, Yuhan Gao, Yuqi Wen, Xuemei Ke, Zejin Ou, Jiaqi Fu, Mingji Cheng, Yun Mo and Qing Chen. Ungulate bocaparvovirus 4 and rodent bocavirus are different genotypes of the same species of virus[J]. Virologica Sinica, 2022, 37(2): 215-222. doi: 10.1016/j.virs.2022.02.002.
Bocaviruses are associated with many human infectious diseases, such as respiratory tract infections, gastroenteritis, and hepatitis. Rats are known to be reservoirs of bocaviruses, including rodent bocavirus and rat bocavirus. Recently, ungulate bocaparvovirus 4, a known porcine bocavirus, has also been found in rats. Thus, investigating bocaviruses in rats is important for determining the origin of the viruses and preventing and controlling their transmission. To the best of our knowledge, no study to date has investigated bocaviruses in the livers of rats. In this report, a total of 624 rats were trapped in southern China between 2014 and 2017. Liver and serum samples from rats were tested for the prevalence of bocaviruses using PCR. Sequences related to ungulate bocaparvovirus 4 and rodent bocavirus were detected in both liver and serum samples. Interestingly, the prevalence of ungulate bocaparvovirus 4 (reference strain:KJ622366.1) was higher than that of rodent bocavirus (reference strain:KY927868.1) in both liver (2.24% and 0.64%, respectively) and serum samples (2.19% and 0.44%, respectively). The NS1 regions of ungulate bocaparvovirus 4 and rodent bocavirus related sequences displayed over 84% and 88% identity at the nucleic acid and amino acid levels, respectively. Furthermore, these sequences had similar genomic structure, genomic features, and codon usage bias, and shared a common ancestor. These viruses also displayed greater adaptability to rats than pigs. Our results suggested that ungulate bocaparvovirus 4 and rodent bocavirus may originate from rats and may be different genotypes of the same bocavirus species.
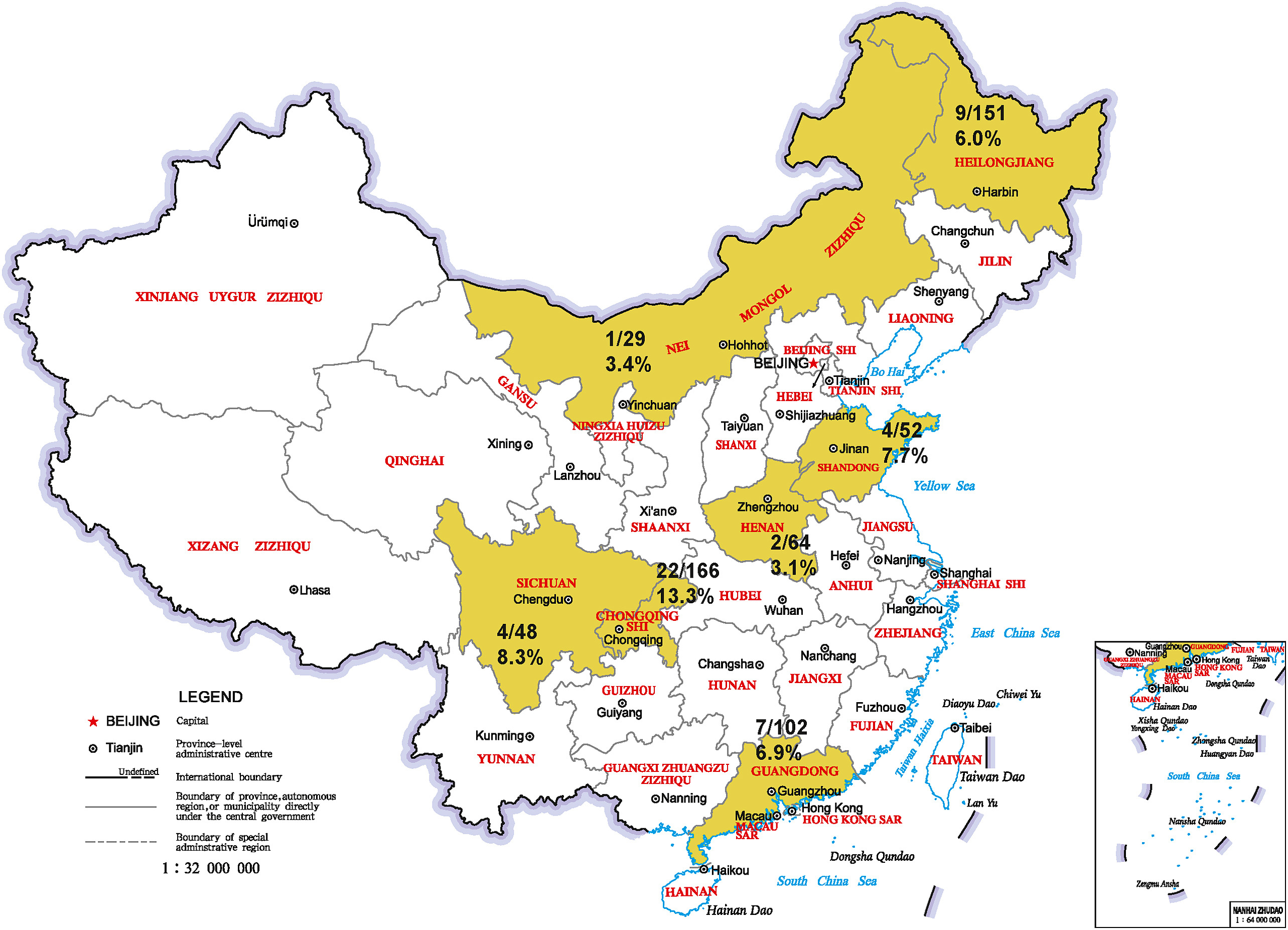
Gang Lu, Chaoxi Chen, Ran Shao, Juan Zhang, Jinghao Li, Siqi Cai, Lintao Zhong, Zhiying Lai, Jiajun Ou, Xin Yin, Guihong Zhang and Shoujun Li. Identification and genetic characterization of bovine hepacivirus in China: A large scale epidemiological study[J]. Virologica Sinica, 2022, 37(2): 223-228. doi: 10.1016/j.virs.2022.02.003.
Bovine hepacivirus (BovHepV) is a novel virus that was recently discovered in Ghana and Germany in 2015. Until now, this virus has been identified in cattle population worldwide and is classified into subtypes A-G. To fully understand the epidemic situation and genetic characteristic of BovHepV in China, a total of 612 cattle serum samples were collected from 20 farms in seven provinces and municipality in China between 2018 and 2020 and were tested for the presence of BovHepV RNA via semi-nested PCR. The results demonstrated that 49 (8.0%) samples were BovHepV RNA-positive. It is noted that BovHepV infection in yak was confirmed for the first time. BovHepV was detected in all the seven provinces, with the positive rate ranging from 3.1% to 13.3%, which indicates a wide geographical distribution pattern of BovHepV in China. Sequencing results revealed that 50 UTR of the 49 field BovHepV strains have a nucleotide similarity of 96.3%-100% between each other and 93.9%- 100% with previously reported BovHepV strains. In addition, genetic analysis identified five critical nucleotide sites in 50 UTR to distinguish different subtypes, which was further verified by genomic sequencing and nucleotide similarity analysis. All the 49 Chinese field BovHepV strains were classified into subtype G and this subtype is only determined in cattle in China currently. This study will provide insights for us to better understand the epidemiology and genetic diversity of BovHepV.
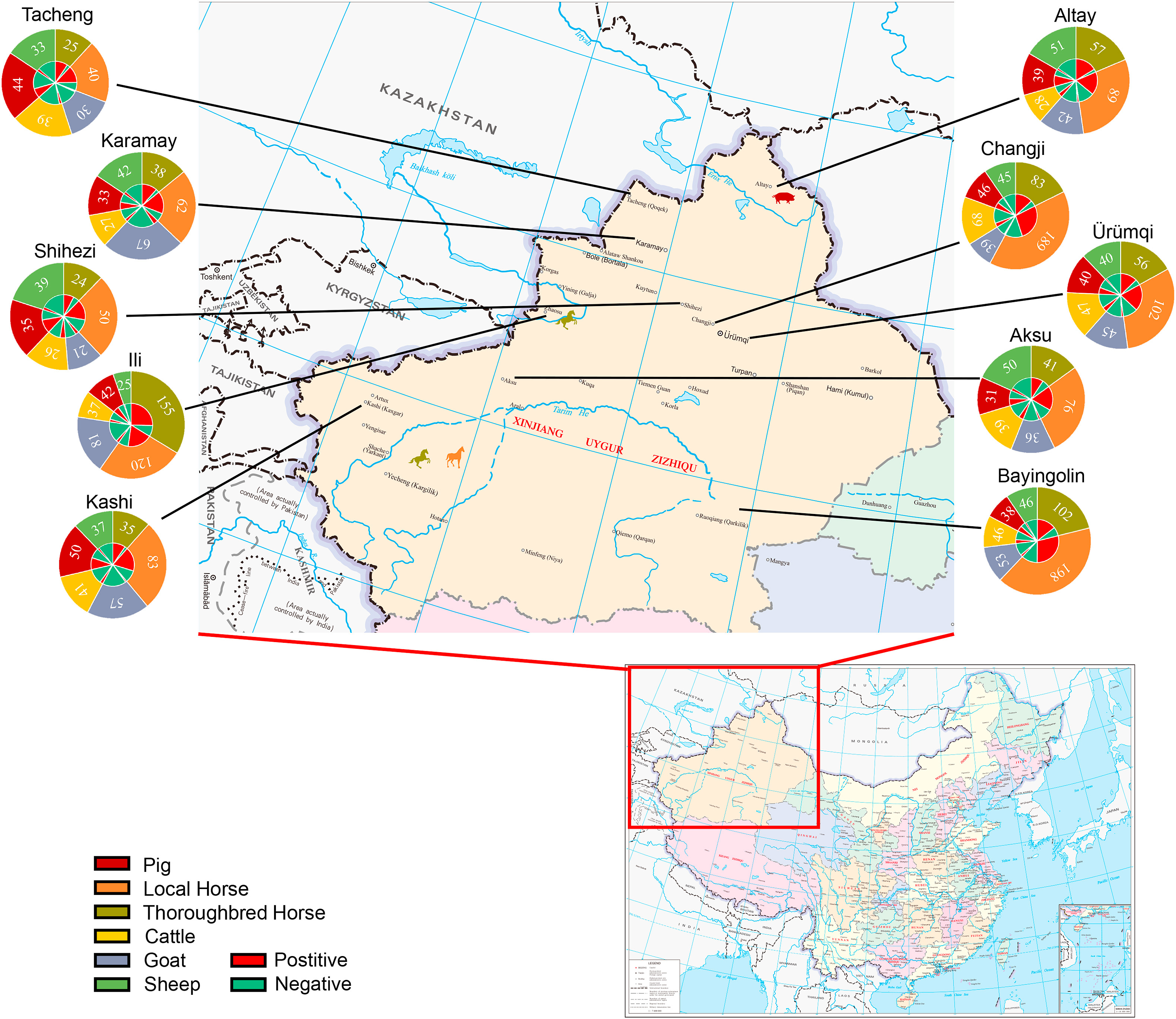
Ning Shi, Xiangshu Qiu, Xinyu Cao, Zhanhai Mai, Xiangyu Zhu, Nan Li, He Zhang, Jinyong Zhang, Zhuoxin Li, Nuerlan Shaya, Huijun Lu and Ningyi Jin. Molecular and serological surveillance of Getah virus in the Xinjiang Uygur Autonomous Region, China, 2017–2020[J]. Virologica Sinica, 2022, 37(2): 229-237. doi: 10.1016/j.virs.2022.02.004.
The Getah virus (GETV), a mosquito-borne RNA virus, is widely distributed in Oceania and Asia. GETV is not the only pathogenic to horses, pigs, cattle, foxes and boars, but it can also cause fever in humans. Since its first reported case in Chinese mainland in 2017, the number of GETV-affected provinces has increased to seventeen till now. Therefore, we performed an epidemiologic investigation of GETV in the Xinjiang region, located in northwestern China, during the period of 2017-2020. ELISA was used to analyze 3299 serum samples collected from thoroughbred horse, local horse, sheep, goat, cattle, and pigs, with thoroughbred horse (74.8%), local horse (67.3%), goat (11.7%), sheep (10.0%), cattle (25.1%) and pigs (51.1%) being positive for anti-GETV antibodies. Interestingly, the neutralizing antibody titer in horses was much higher than in other species. Four samples from horses and pigs were positive for GETV according to RT-PCR. Furthermore, from the serum of a local horse, we isolated GETV which was designated as strain XJ-2019-07, and determined its complete genome sequence. From the phylogenetic relationships, it belongs to the Group III lineage. This is the first evidence of GETV associated to domestic animals in Xinjiang. Overall, GETV is prevalent in Xinjiang and probably has been for several years. Since no vaccine against GETV is available in China, detection and monitoring strategies should be improved in horses and pigs, especially imported and farmed, in order to prevent economic losses.
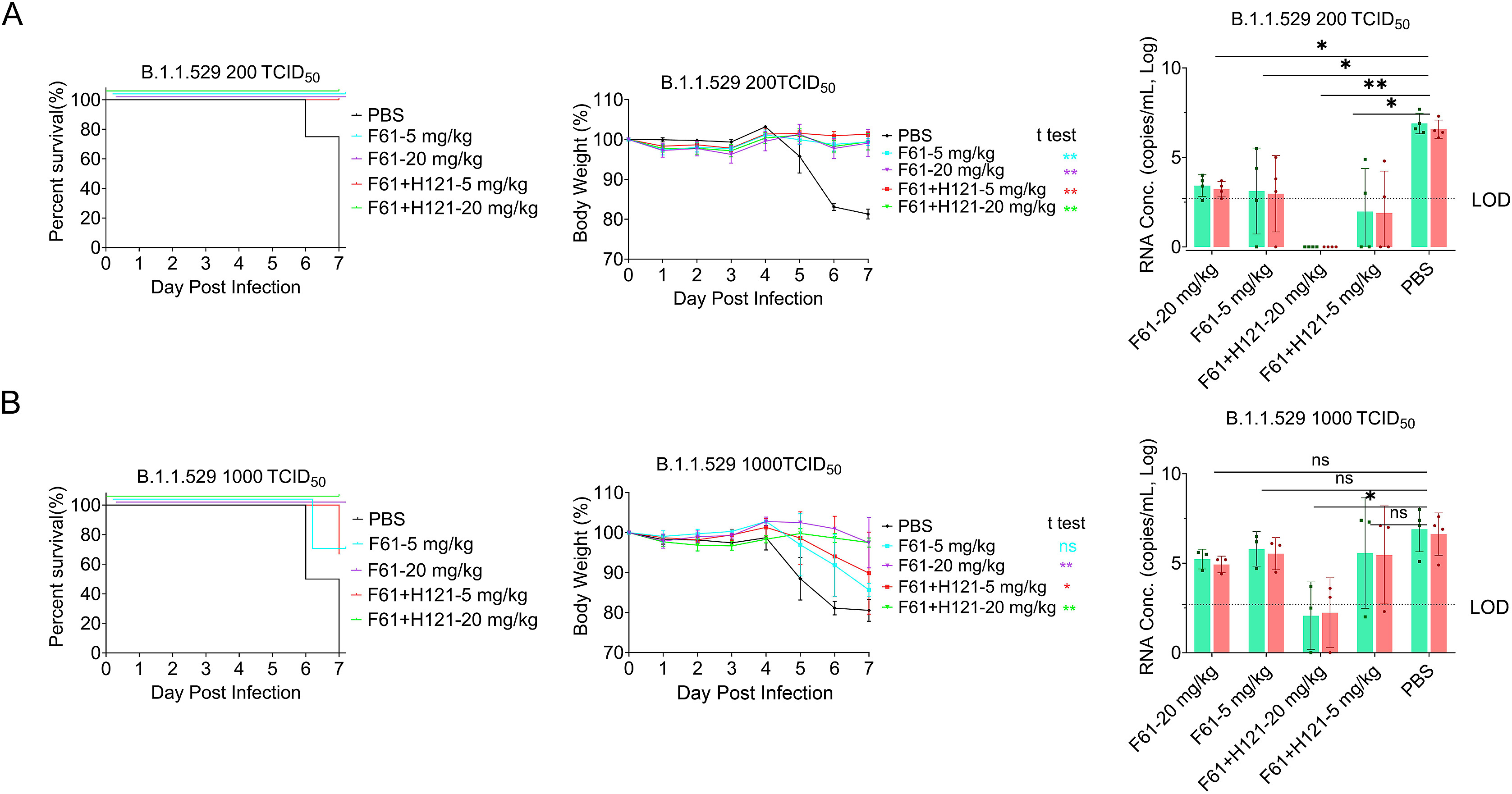
Multiple new variants of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have constantly emerged, as the delta and omicron variants, which have developed resistance to currently gained neutralizing antibodies. This highlights a critical need to discover new therapeutic agents to overcome the variants mutations. Despite the availability of vaccines against coronavirus disease 2019 (COVID-19), the use of broadly neutralizing antibodies has been considered as an alternative way for the prevention or treatment of SARS-CoV-2 variants infection. Here, we show that the nasal delivery of two previously characterized broadly neutralizing antibodies (F61 and H121) protected K18-hACE2 mice against lethal challenge with SARS-CoV-2 variants. The broadly protective efficacy of the F61 or F61/F121 cocktail antibodies was evaluated by lethal challenge with the wild strain (WIV04) and multiple variants, including beta (B.1.351), delta (B.1.617.2), and omicron (B.1.1.529) at 200 or 1000 TCID50, and the minimum antibody administration doses (5-1.25 mg/kg body weight) were also evaluated with delta and omicron challenge. Fully prophylactic protections were found in all challenged groups with both F61 and F61/H121 combination at the administration dose of 20 mg/kg body weight, and corresponding mice lung viral RNA showed negative, with almost all alveolar septa and cavities remaining normal. Furthermore, low-dose antibody treatment induced significant prophylactic protection against lethal challenge with delta and omicron variants, whereas the F61/H121 combination showed excellent results against omicron infection. Our findings indicated the potential use of broadly neutralizing monoclonal antibodies as prophylactic and therapeutic agent for protection of current emerged SARS-CoV-2 variants infection.
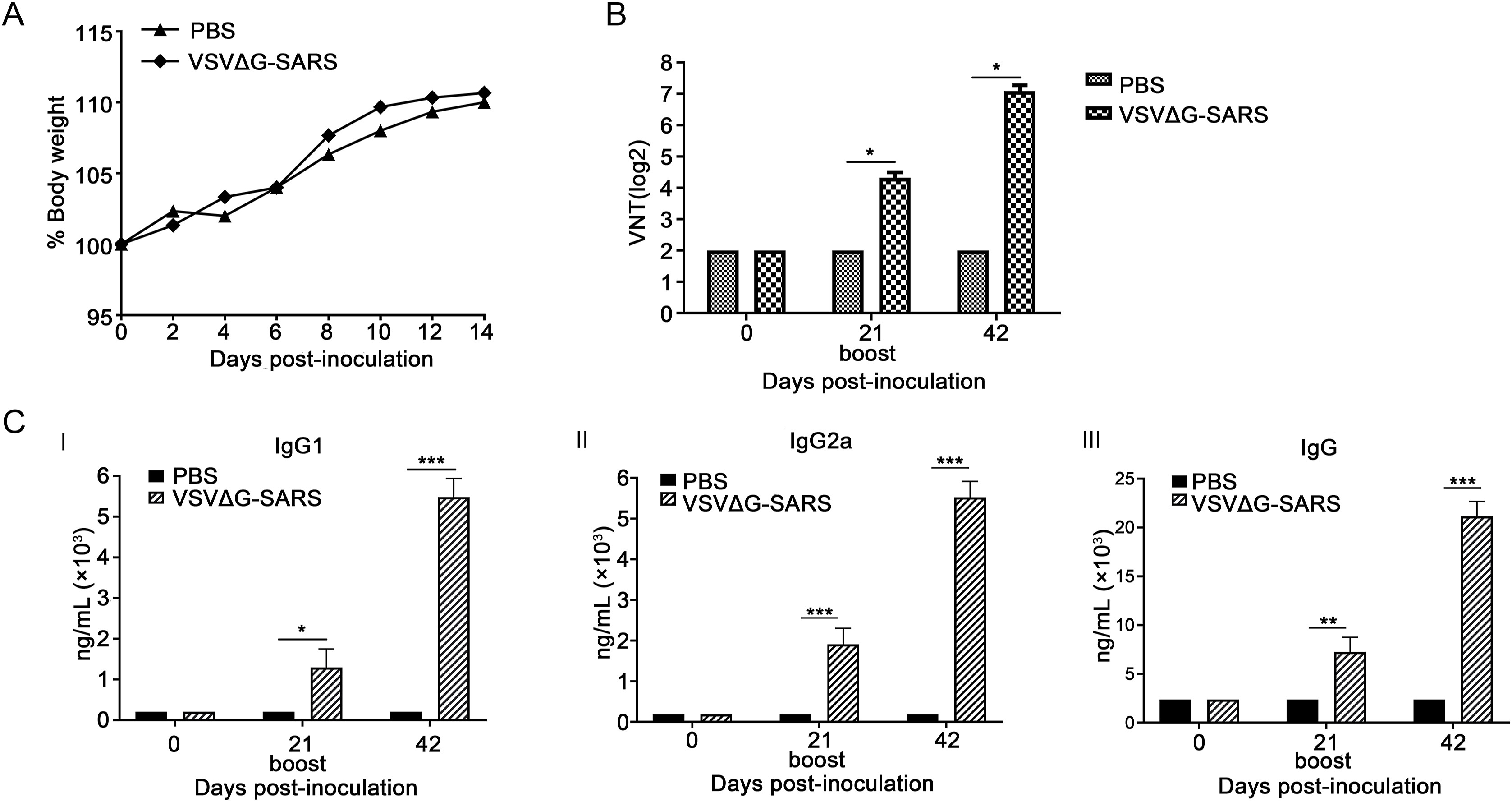
Dan Shan, Xiaoyan Tang, Renqiang Liu, Dan Pan, Xijun Wang, Jinying Ge, Zhiyuan Wen and Zhigao Bu. Immunogenicity of a recombinant VSV-Vectored SARS-CoV vaccine induced robust immunity in rhesus monkeys after single-dose immunization[J]. Virologica Sinica, 2022, 37(2): 248-255. doi: 10.1016/j.virs.2022.01.002.
Severe acute respiratory syndrome (SARS) is a highly contagious zoonotic disease caused by SARS coronavirus (SARS-CoV). Since its outbreak in Guangdong Province of China in 2002, SARS has caused 8096 infections and 774 deaths by December 31st, 2003. Although there have been no more SARS cases reported in human populations since 2004, the recent emergence of a novel coronavirus disease (COVID-19) indicates the potential of the recurrence of SARS and other coronavirus disease among humans. Thus, developing a rapid response SARS vaccine to provide protection for human populations is still needed. Spike (S) protein of SARS-CoV can induce neutralizing antibodies, which is a pivotal immunogenic antigen for vaccine development. Here we constructed a recombinant chimeric vesicular stomatitis virus (VSV) VSVΔG-SARS, in which the glycoprotein (G) gene is replaced with the SARS-CoV S gene. VSVΔG-SARS maintains the bullet-like shape of the native VSV, with the heterogeneous S protein incorporated into its surface instead of G protein. The results of safety trials revealed that VSVΔG-SARS is safe and effective in mice at a dose of 1×106 TCID50. More importantly, only a single-dose immunization of 2×107 TCID50 can provide high-level neutralizing antibodies and robust T cell responses to non-human primate animal models. Thus, our data indicate that VSVΔG-SARS can be used as a rapid response vaccine candidate. Our study on the recombinant VSV-vectored SARS-CoV vaccines can accumulate experience and provide a foundation for the new coronavirus disease in the future.
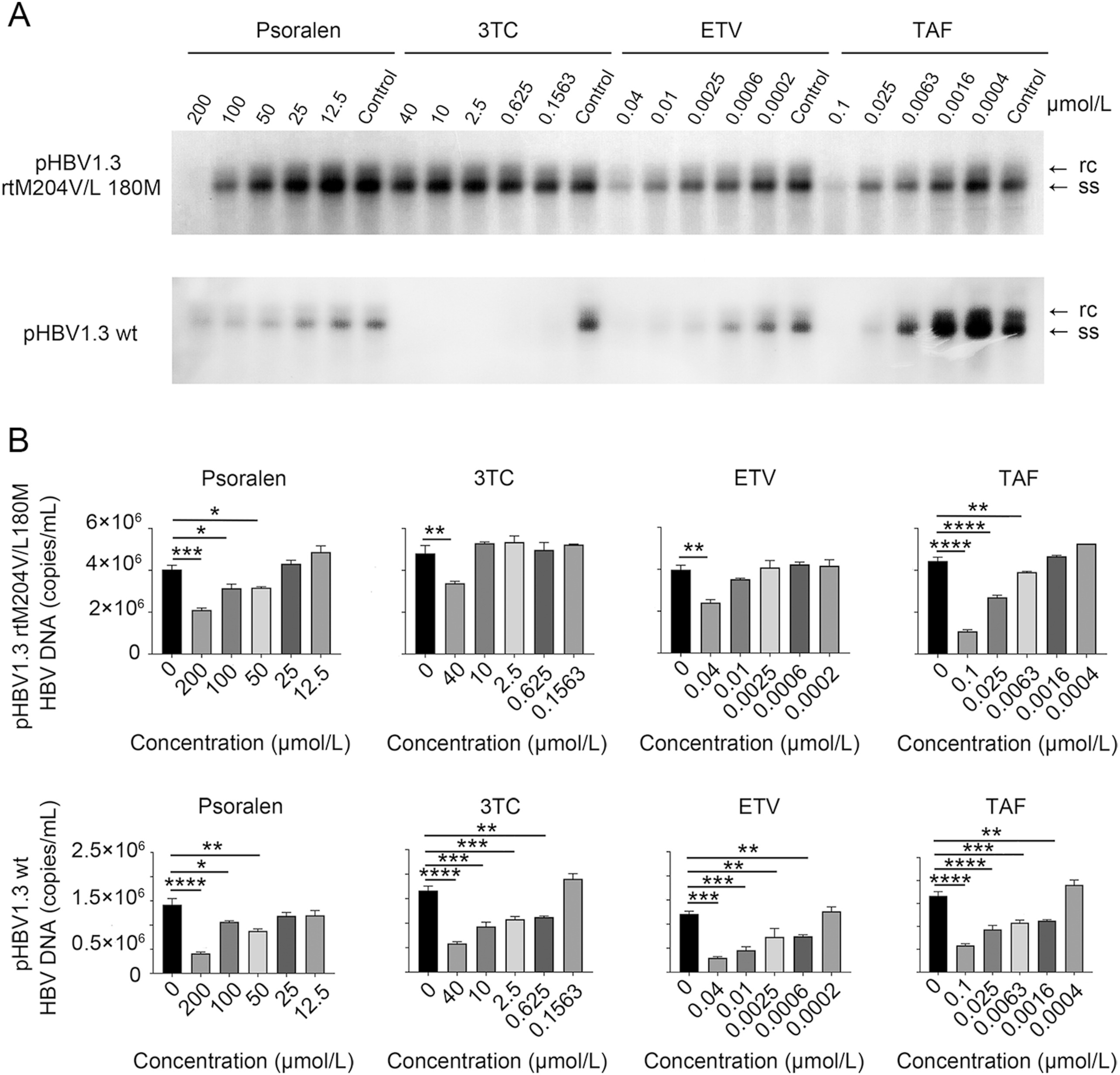
Xinna Ma, Heng Li, Ying Gong, Feifei Liu, Xiankun Tong, Fenghua Zhu, Xiaoqian Yang, Li Yang and Jianping Zuo. Psoralen inhibits hepatitis B viral replication by down-regulating the host transcriptional machinery of viral promoters[J]. Virologica Sinica, 2022, 37(2): 256-265. doi: 10.1016/j.virs.2022.01.027.
The hepatitis B virus (HBV) is a global public health challenge due to its highly contagious nature. It is estimated that almost 300 million people live with chronic HBV infection annually. Although nucleoside analogs markedly reduce the risk of liver disease progression, the analogs do not fully eradicate the virus. As such, new treatment options and drugs are urgently needed. Psoralen is a nourishing monomer of Chinese herb and is known to inhibit virus replication and inactivate viruses. In this study, we evaluated the potential of psoralen as an anti-HBV agent. Quantitative PCR and Southern blot analysis revealed that psoralen inhibited HBV replication in HepG2.2.15 cells in a concentration-dependent manner. Moreover, psoralen was also active against the 3TC/ETV-dual-resistant HBV mutant. Further investigations revealed that psoralen suppressed both HBV RNA transcription and core protein expression. The transcription factor FOXO1, a known target for PGC1α co-activation, binds to HBV pre-core/core promoter enhancer II region and activates HBV RNA transcription. Co-immunoprecipitation showed that psoralen suppressed the expression of FOXO1, thereby decreasing the binding of FOXO1 co-activator PGC1α to the HBV promoter. Overall, our results demonstrate that psoralen suppresses HBV RNA transcription by down-regulating the expression of FOXO1 resulting in a reduction of HBV replication.
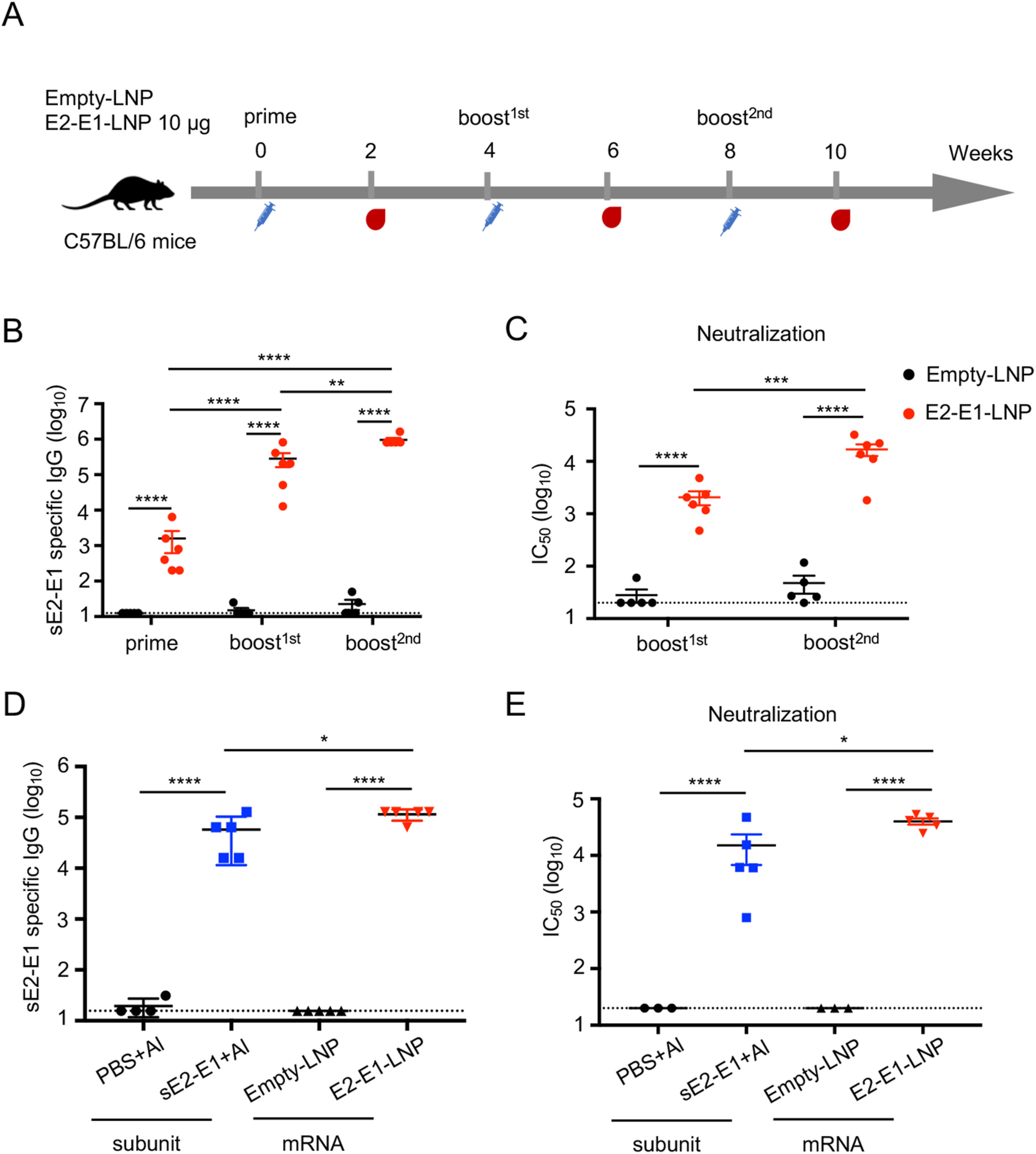
Ningning Ge, Jin Sun, Zhihua Liu, Jiayi Shu, Huimin Yan, Zhihua Kou, Yu Wei and Xia Jin. An mRNA vaccine encoding Chikungunya virus E2-E1 protein elicits robust neutralizing antibody responses and CTL immune responses[J]. Virologica Sinica, 2022, 37(2): 266-276. doi: 10.1016/j.virs.2022.01.032.
Arthropod-borne chikungunya virus (CHIKV) infection can cause a debilitating arthritic disease in human. However, there are no specific antiviral drugs and effective licensed vaccines against CHIKV available for clinical use. Here, we developed an mRNA-lipid nanoparticle (mRNA-LNP) vaccine expressing CHIKV E2-E1 antigen, and compared its immunogenicity with soluble recombinant protein sE2-E1 antigen expressed in S2 cells. For comparison, we first showed that recombinant protein antigens mixed with aluminum adjuvant elicit strong antigen-specific humoral immune response and a moderate cellular immune response in C57BL/6 mice. Moreover, sE2-E1 vaccine stimulated 12-23 folds more neutralizing antibodies than sE1 vaccine and sE2 vaccine. Significantly, when E2-E1 gene was delivered by an mRNA-LNP vaccine, not only the better magnitude of neutralizing antibody responses was induced, but also greater cellular immune responses were generated, especially for CD8+ T cell responses. Moreover, E2-E1-LNP induced CD8+ T cells can perform cytotoxic effect in vivo. Considering its better immunogenicity and convenience of preparation, we suggest that more attention should be placed to develop CHIKV E2-E1-LNP mRNA vaccine.
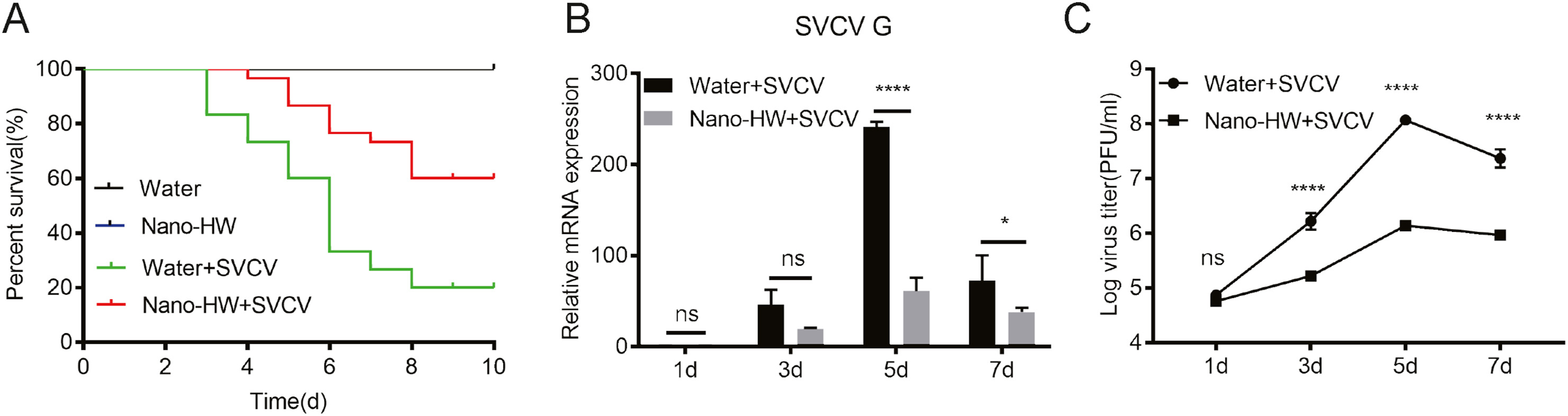
Chen Li, Yiran Cao, Fukuda Kohei, Haihong Hao, Guiqing Peng, Can Cheng and Jing Ye. Nano-bubble hydrogen water: An effective therapeutic agent against inflammation related disease caused by viral infection in zebrafish model[J]. Virologica Sinica, 2022, 37(2): 277-283. doi: 10.1016/j.virs.2022.01.023.
Since the anti-inflammatory effect of hydrogen has been widely known, it was supposed that hydrogen could suppress tissue damage by inhibiting virus-related inflammatory reactions. However, hydrogen is slightly soluble in water, which leads to poor effect of oral hydrogen-rich water therapy. In this study, the nano-bubble hydrogen water (nano-HW) (about 0.7 ppm) was prepared and its therapeutic effect against viral infection was investigated by utilizing spring viraemia of carp virus (SVCV)-infected zebrafish as model. Three-month-old zebrafish were divided into nano-HW treatment-treated group and aquaculture water treated group (control group). The results revealed that the cumulative mortality rate of SVCV-infected zebrafish was reduced by 40% after treatment with nano-bubble hydrogen water, and qRT-PCR results showed that SVCV replication was significantly inhibited. Histopathological examination staining showed that SVCV infection caused tissue damage was greatly alleviated after treatment with nano-bubble hydrogen water. Futhermore, SVCV infection caused reactive oxygen species (ROS) accumulation was significantly reduced upon nano-HW treatment. The level of proinflammatory cytokines IL-1β, IL-8, and TNF-α was remarkably reduced in the nano-HW-treated group in vivo and in vitro. Taken together, our data demonstrated for the first time that nano-HW could inhibit the inflammatory response caused by viral infection in zebrafish, which suggests that nano-HW can be applied to antiviral research,and provides a novel therapeutic strategy for virus-caused inflammation related disease.
Subgroup J avian leukosis virus (ALV-J) is a highly oncogenic retrovirus that has been devastating the global poultry industry since the late 1990s. The major infection model of ALV-J is vertical transmission, which is responsible for the congenital infection of progeny from generation to generation. Increasing evidence has suggested that extracellular vesicles (EVs) derived from virus-infected cells or biological fluids have been thought to be vehicles of transmission for viruses. However, the role of EVs in infection and transmission of ALV-J remains obscure. In the present study, semen extracellular vesicles (SE) were isolated and purified from ALV-J-infected rooster seminal plasma (SE-ALV-J), which was shown to contain ALV-J genomic RNA and partial viral proteins, as determined by RNA sequencing, reverse transcription-quantitative PCR and Western blotting. Furthermore, SE-ALV-J was proved to be able to transmit ALV-J infection to host cells and establish productive infection. More importantly, artificial insemination experiments showed that SE-ALV-J transmitted ALV-J infection to SPF hens, and subsequently mediated vertical transmission of ALV-J from the SPF hens to the progeny chicks. Taken together, the results of the present study suggested that ALV-J utilized host semen extracellular vesicles as a novel means for vertical transmission, enhancing our understanding on mechanisms underlying ALV-J transmission.
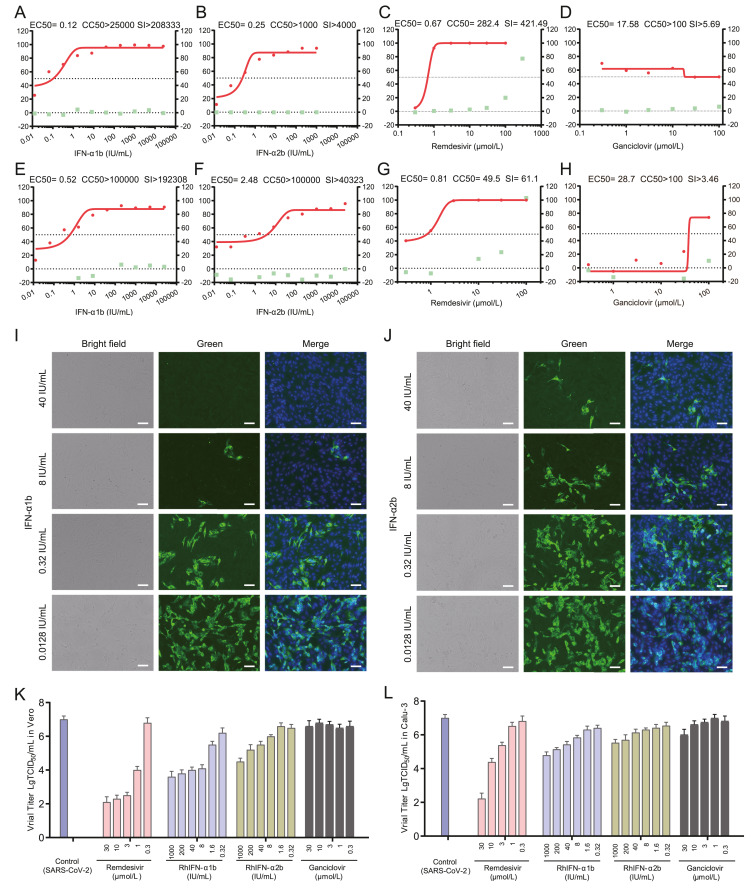
Danrong Shi, Keda Chen, Xiangyun Lu, Linfang Cheng, Tianhao Weng, Fumin Liu, Nanping Wu, Lanjuan Li and Hangping Yao. Recombinant human interferon-α1b inhibits SARS-CoV-2 better than interferon-α2b in vitro[J]. Virologica Sinica, 2022, 37(2): 295-298. doi: 10.1016/j.virs.2022.01.031.
Highlights 1) A comprehensive evaluation method for anti-SARS-CoV-2 drugs was established based on RT-qPCR, TCID50 method, and immunofluorescence. 2) A significant antiviral effect of rHuIFN-α1b was shown with EC50=0.12 IU/mL in Vero cells and EC50=0.52 IU/mL in Calu-3 cells, which was better than rHuIFN-α2b (EC50=0.25 IU/mL in Vero cells and EC50=2.48 IU/mL in Calu-3 cells). 3) rHuIFN-α1b has a good potential in the application of anti-COVID-19 therapy.
Ge Gao, Xue Hu, Yiwu Zhou, Juhong Rao, Xiaoyu Zhang, Yun Peng, Jiaxuan Zhao, Yanfeng Yao, Kunpeng Liu, Mengying Liang, Hang Liu, Fei Deng, Han Xia, Chao Shan and Zhiming Yuan. Infection and pathogenesis of the Delta variant of SARS-CoV-2 in Rhesus macaque[J]. Virologica Sinica, 2022, 37(2): 299-302. doi: 10.1016/j.virs.2022.02.001.
Highlights 1. Delta variant of SARS-CoV-2 can effectively infect the Rhesus macaque. 2. The Delta variant grows faster than the early strain isolated from Wuhan in late 2019. 3. The shedding pattern, viral load and disease severity of Delta variant are similar with the early strain isolated from Wuhan in late 2019. 4. This study supports the attributed rapid disease spread of the Delta variant.
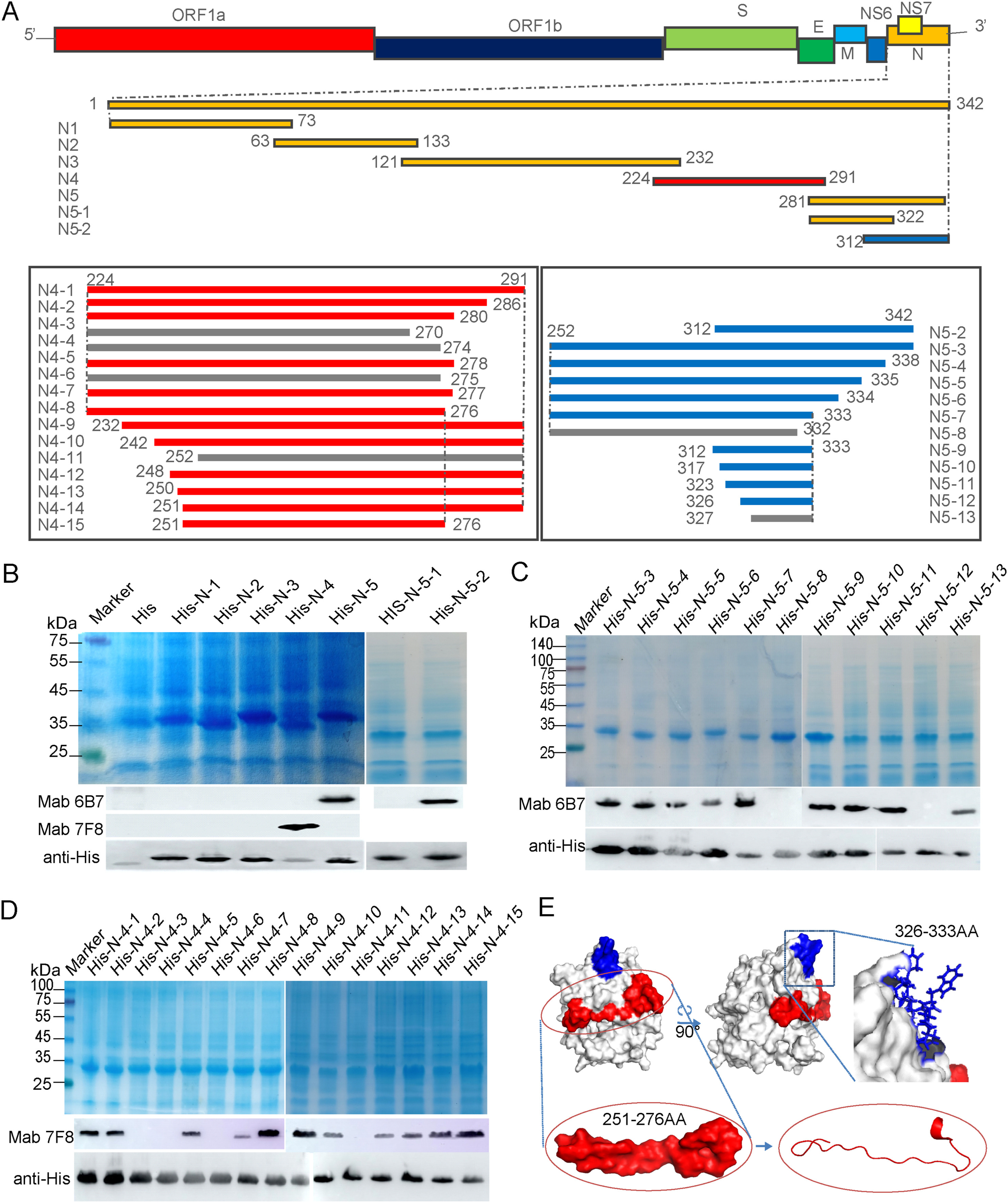
Haojie Ren, Xiaoguang Yan, Lintao Liu, Yixuan Zhang, Qianqian Li, Xiumei Li and Hui Hu. Identification of two novel B-cell epitopes on the nucleocapsid protein of porcine deltacoronavirus[J]. Virologica Sinica, 2022, 37(2): 303-306. doi: 10.1016/j.virs.2022.01.025.
Highlights 1. Two monoclonal antibodies against newly emerged porcine deltacoronavirus nucleocapsid protein were prepared. 2. The epitopes that these two monoclonal antibodies recognized on nucleocapsid protein were identified. 3. The monoclonal antibody 6B7 recognized a linear epitope of N protein, while the 7F8 recognized a conformational epitope. 4. Conservation of the identified epitopes between different coronaviruses was analyzed.
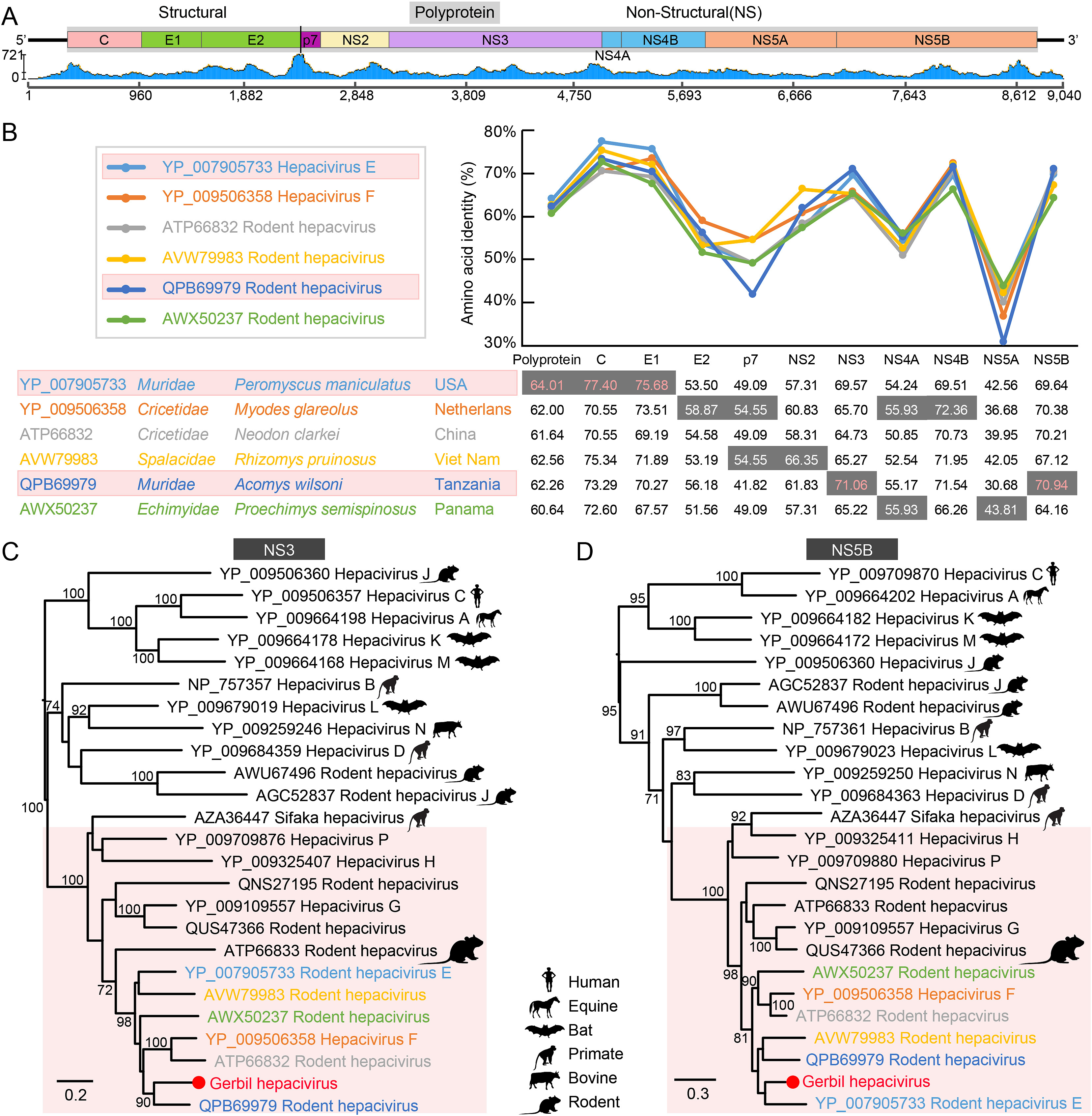
Cui-hong An, Juan Li, Yi-ting Wang, Shou-min Nie, Wen-hui Chang, Hong Zhou, Lin Xu, Yang-xin Sun, Wei-feng Shi and Ci-xiu Li. Identification of a novel hepacivirus in Mongolian gerbil (Meriones unguiculatus) from Shaanxi, China[J]. Virologica Sinica, 2022, 37(2): 307-310. doi: 10.1016/j.virs.2022.01.016.
Highlights: 1. The first hepacivirus detected in Mongolian gerbils from a plague zones in China. 2. A novel hepacivirus closely related to hepacivirus E and F. 3. Mongolian gerbils could be a potential animal model for hepacivirus pathogenicity. 4. Extending the genetic diversity and host range of hepaciviruses.
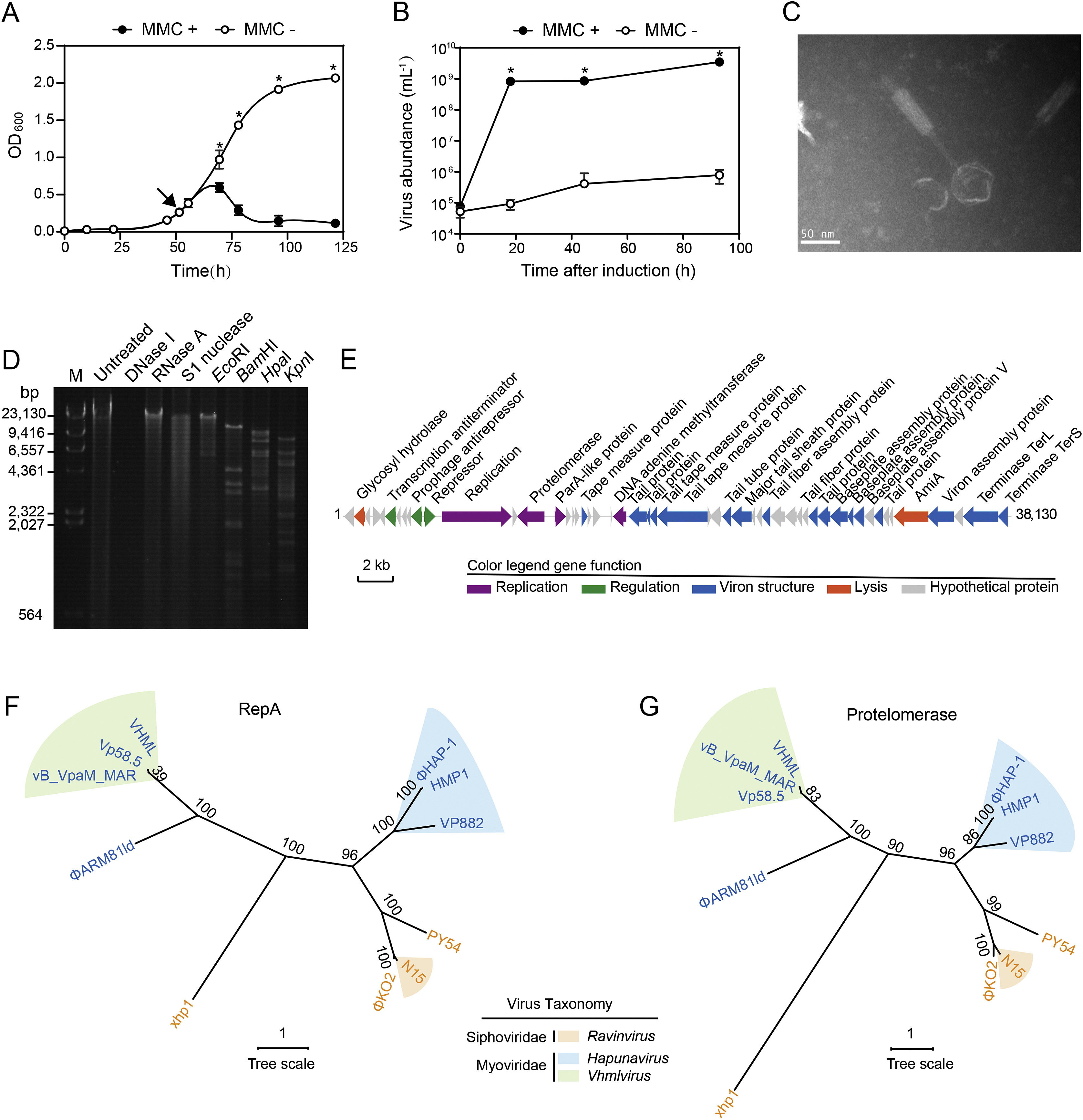
Yali Hao, Siyuan Wang, Mujie Zhang, Qingxue Tang, Canxing Meng, Liping Wang, Qilian Fan, Yaxian Yan, Xiang Xiao and Huahua Jian. Isolation and characterization of a novel linear-plasmid phage from the sediment of the Mariana Trench[J]. Virologica Sinica, 2022, 37(2): 311-313. doi: 10.1016/j.virs.2022.01.022.
Highlights 1. HMP1 is the first bacteriophage that was isolated from hadal sediment, the water depth at which HMP1 was isolated is the highest on record up to now. 2. The isolation of HMP1 extends the habitat of linear plasmid phages from the surface to the deep ocean. 3. The genomic and morphological features of HMP1 provide hints with regard to the vertical exchange of viral communities in the ocean. 4. HMP1 and Halomonas sp. MT08-1 contribute a useful phage-host system for in-depth analysis of the life strategy of viruses and their interactions with bacterial hosts in extreme hadal environments.
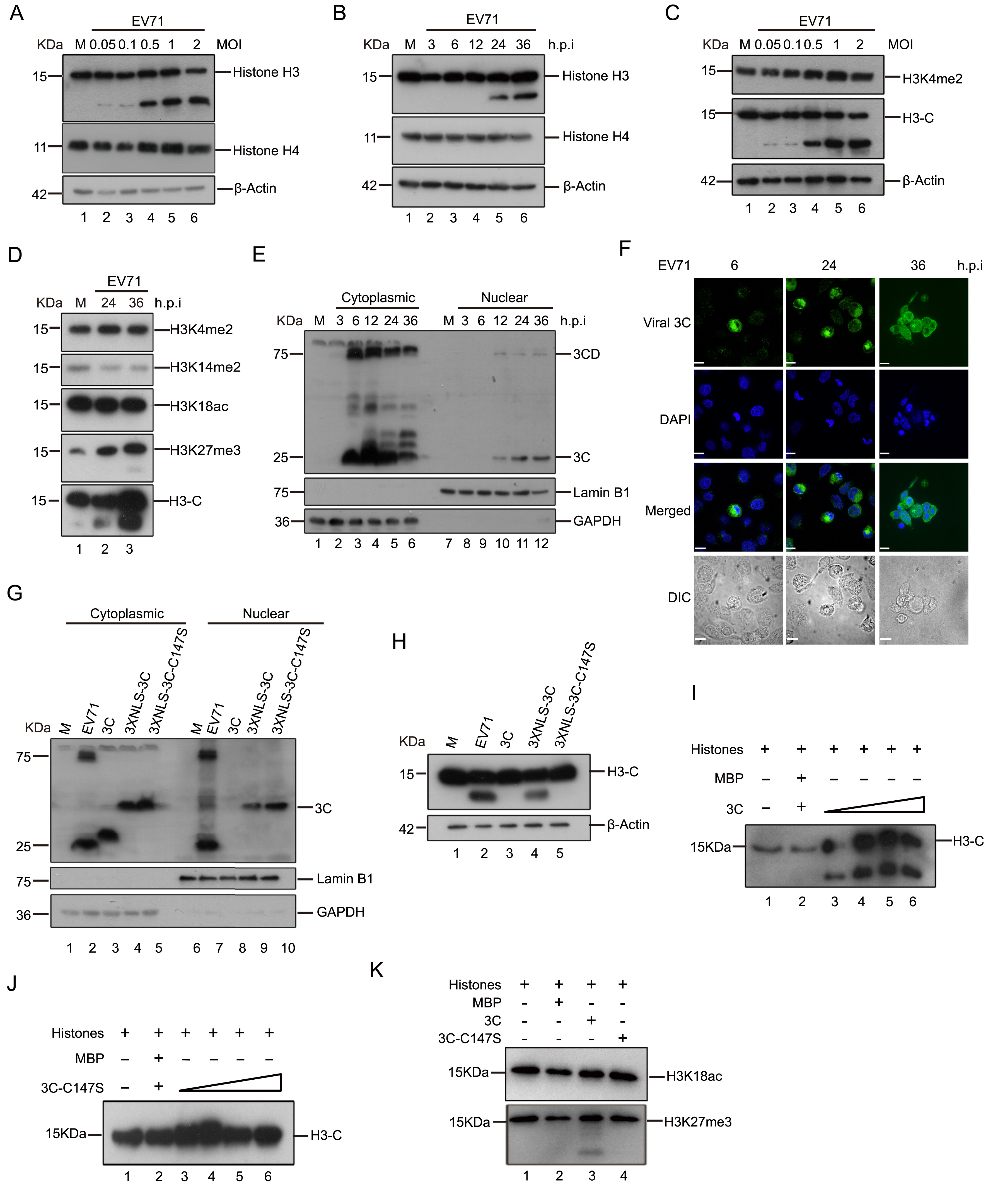
Meng Miao, Gang Deng, Xiaobei Xiong, Yang Qiu, Wenda Huang, Meng Yuan, Fei Yu, Shimei Bai, Xi Zhou and Xiaolu Zhao. Enterovirus 71 3C proteolytically processes the histone H3 N-terminal tail during infection[J]. Virologica Sinica, 2022, 37(2): 314-317. doi: 10.1016/j.virs.2022.02.006.
Highlights 1. The N-terminal tail of histone H3 is specifically cleaved during EV71 infection. 2. Viral protease 3C is identified as a protease responsible for proteolytically processing the N-terminal H3 tail. 3. Our finding reveals a new epigenetic regulatory mechanism for Enterovirus 71 in virus-host interactions.
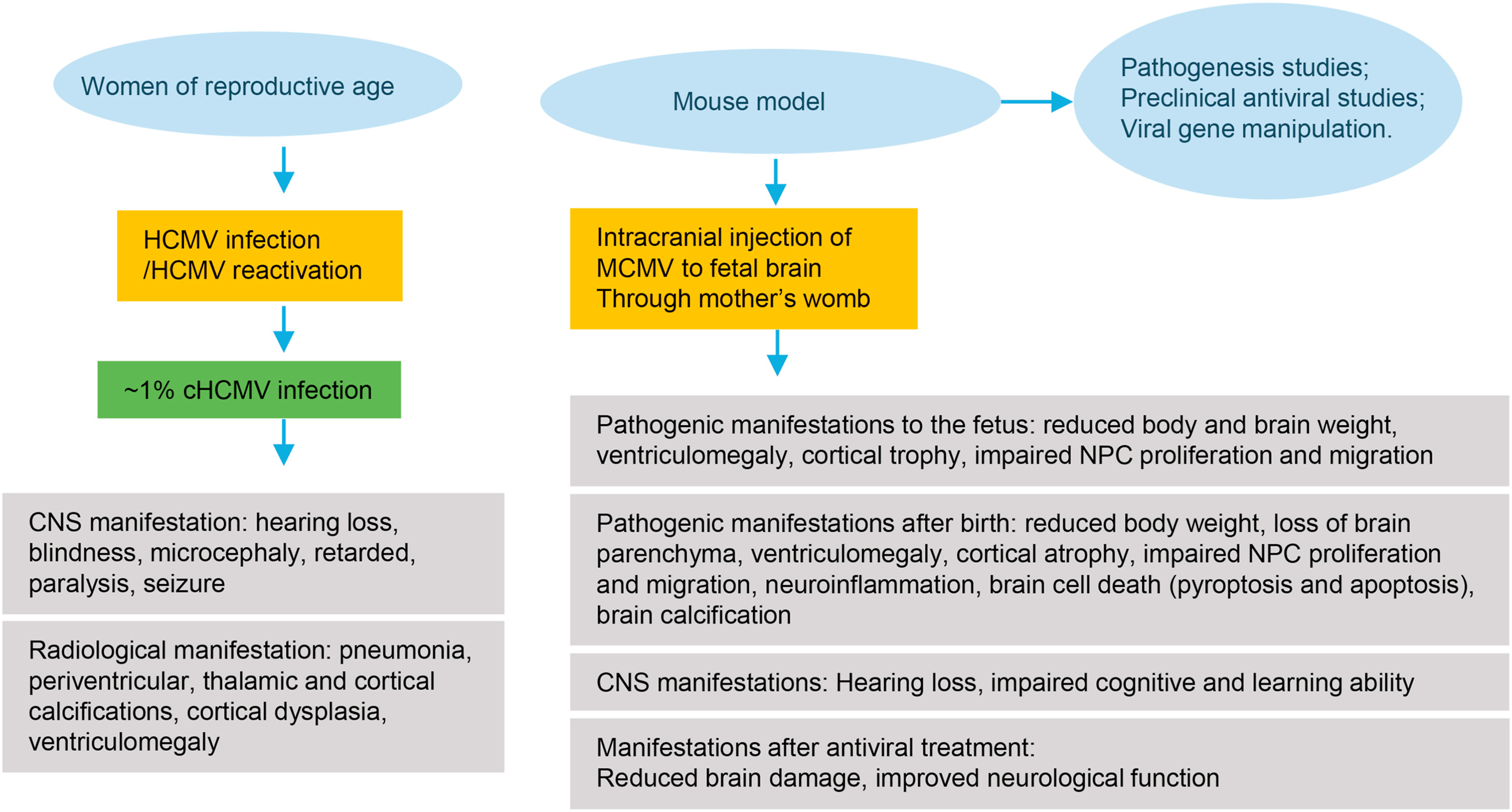
Najealicka Armstrong and Qiyi Tang. Congenital cytomegalovirus infection and advances in murine models of neuropathogenesis[J]. Virologica Sinica, 2022, 37(2): 318-320. doi: 10.1016/j.virs.2022.04.007.
Highlights · Congenital human cytomegalovirus (CMV) infection causes severe neuropathogenesis. · Murine CMV failed to break through the placental barrier to transmit to fetus. · Zhou et al. established a novel mouse system to model congenital HCMV infection. · The mouse CMV system by Zhou et al can be used for drug screening.