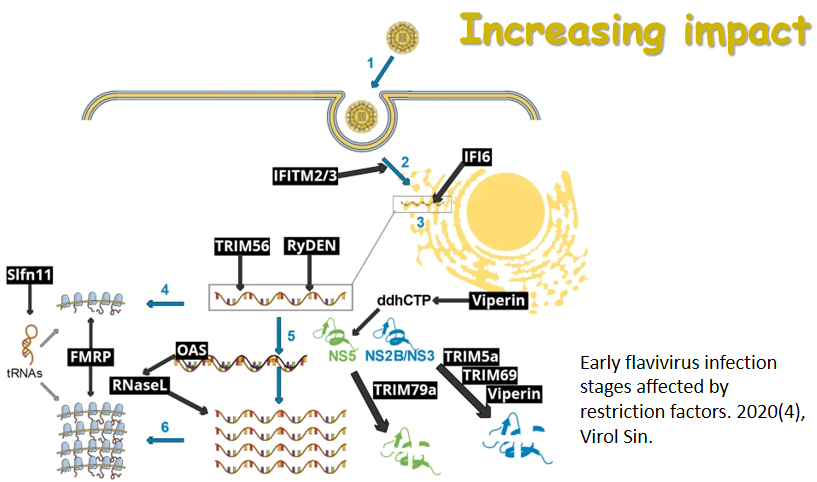
Viral replication is largely host-dependent, with viruses hijacking cellular functions to facilitate their replication, while hosts develop barriers to fight the infection. Cellular restriction factors are early lines of defense against viral replication and spread by interfering with various critical steps in the viral replication cycle. However, viruses have evolved specific mechanisms to overcome these host barriers through adaptation. The battle between the viruses and restriction factors is actually a natural part of the viral replication process. In this issue, Wang et al. systematically reviewed the current literature on the interactions between various cellular restriction factors and the equine infectious anemia virus (EIAV), an animal lentivirus model for HIV/AIDS research. The general functions, targeted key steps, viral antagonists, and restriction mechanisms of cellular restriction factors during the lentiviral replication cycle were summarized. Different strategies between EIAV and HIV-1 against cellular restriction factors were comparatively analyzed. See page 485-496 for the review article.
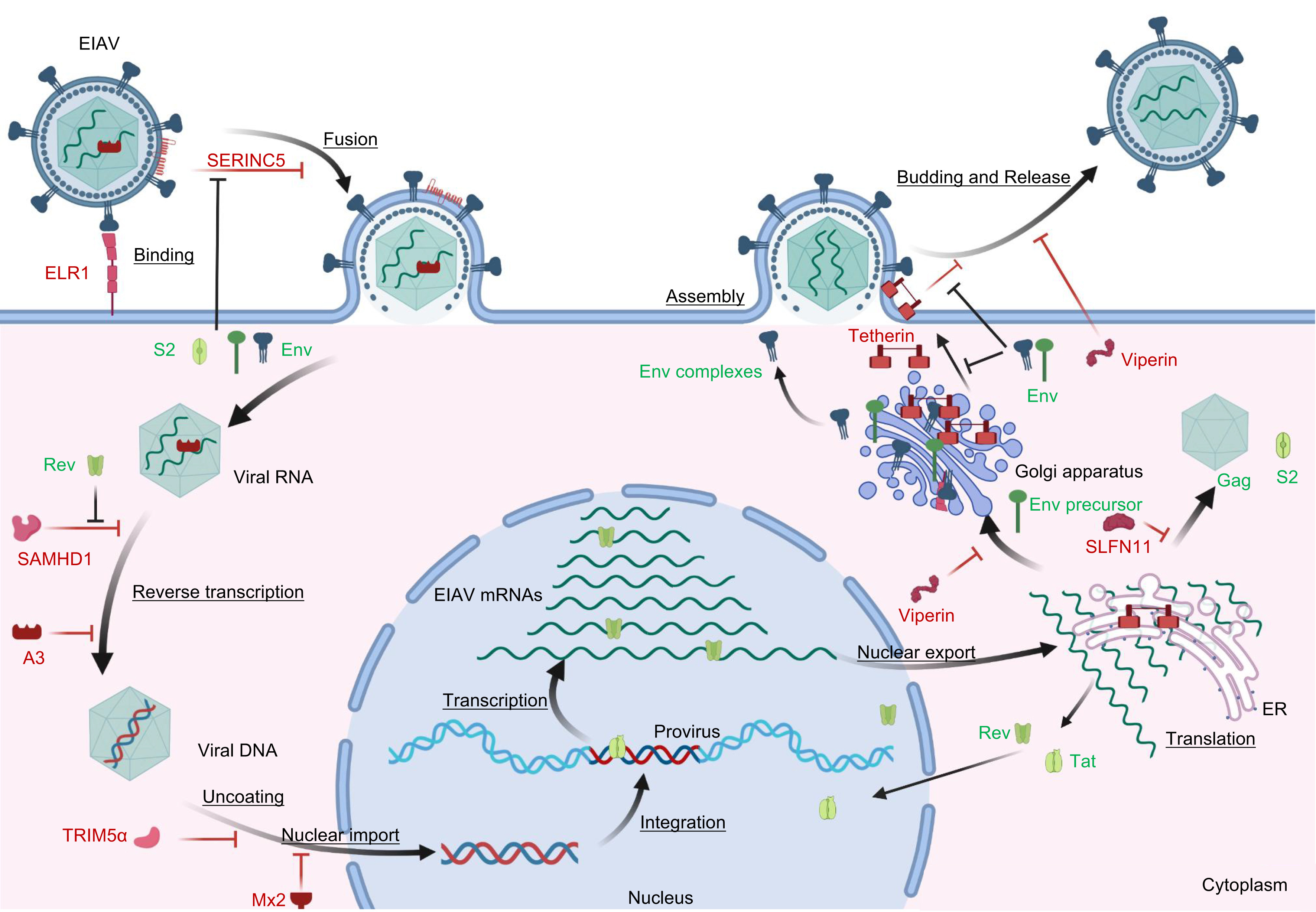
Equine infectious anemia virus (EIAV) is a member of the lentivirus genus in the Retroviridae family and is considered an animal model for HIV/AIDS research. An attenuated EIAV vaccine, which was successfully developed in the 1970s by classical serial passage techniques, is the first and only lentivirus vaccine that has been widely used to date. Restriction factors are cellular proteins that provide an early line of defense against viral replication and spread by interfering with various critical steps in the viral replication cycle. However, viruses have evolved specific mechanisms to overcome these host barriers through adaptation. The battle between the viruses and restriction factors is actually a natural part of the viral replication process, which has been well studied in human immunodeficiency virus type 1 (HIV-1). EIAV has the simplest genome composition of all lentiviruses, making it an intriguing subject for understanding how the virus employs its limited viral proteins to overcome restriction factors. In this review, we summarize the current literature on the interactions between equine restriction factors and EIAV. The features of equine restriction factors and the mechanisms by which the EIAV counteract the restriction suggest that lentiviruses employ diverse strategies to counteract innate immune restrictions. In addition, we present our insights on whether restriction factors induce alterations in the phenotype of the attenuated EIAV vaccine.
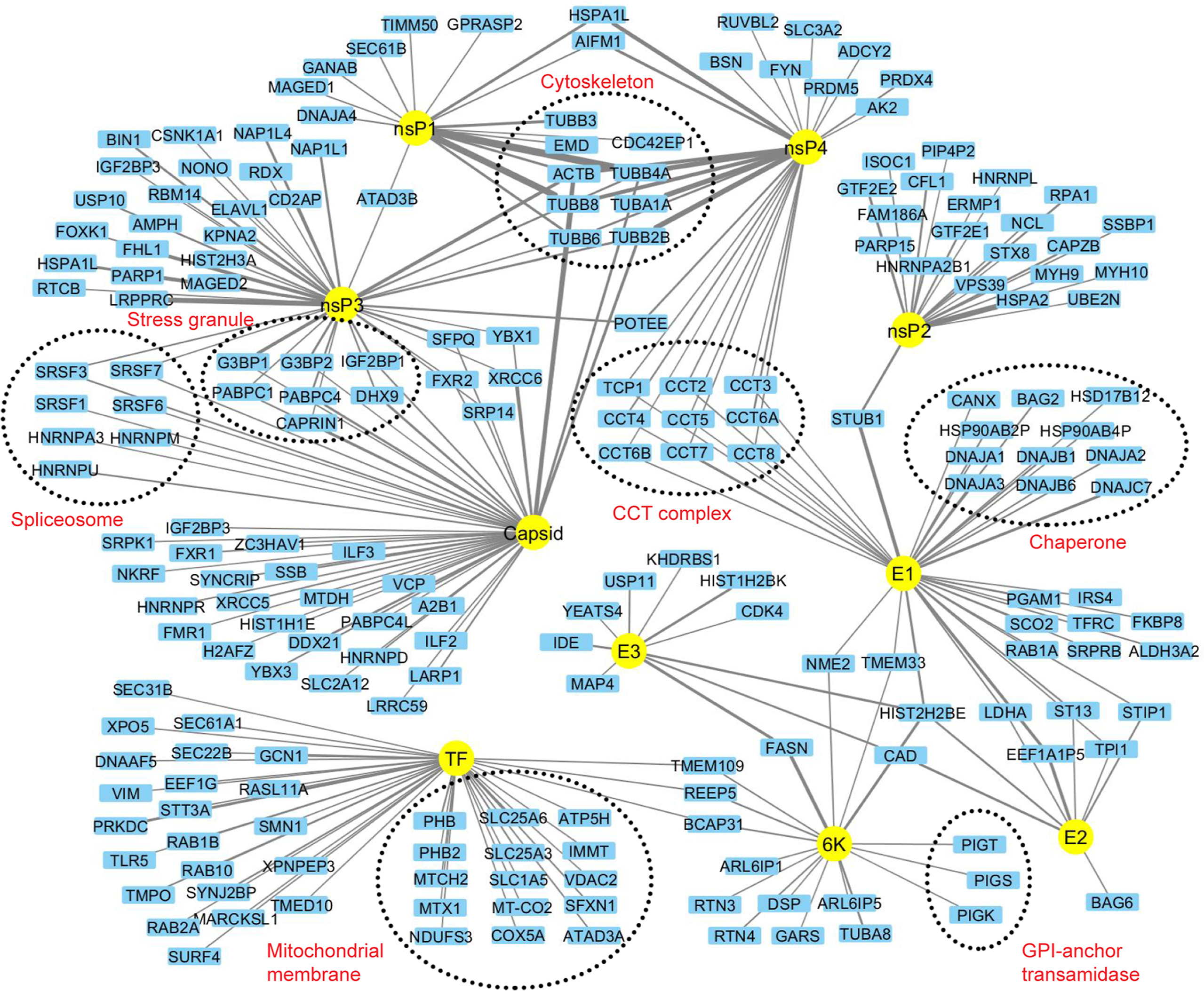
Chikungunya virus (CHIKV) is a re-emerging mosquito-transmitted RNA virus causing joint and muscle pain. To better understand how CHIKV rewires the host cell and usurps host cell functions, we generated a systematic CHIKV-human protein-protein interaction map and revealed several novel connections that will inform further mechanistic studies. One of these novel interactions, between the viral protein E1 and STIP1 homology and U-box containing protein 1 (STUB1), was found to mediate ubiquitination of E1 and degrade E1 through the proteasome. Capsid associated with G3BP1, G3BP2 and AAA+ ATPase valosin-containing protein (VCP). Furthermore, VCP inhibitors blocked CHIKV infection, suggesting VCP could serve as a therapeutic target. Further work is required to fully understand the functional consequences of these interactions. Given that CHIKV proteins are conserved across alphaviruses, many virus-host protein-protein interactions identified in this study might also exist in other alphaviruses. Construction of interactome of CHIKV provides the basis for further studying the function of alphavirus biology.
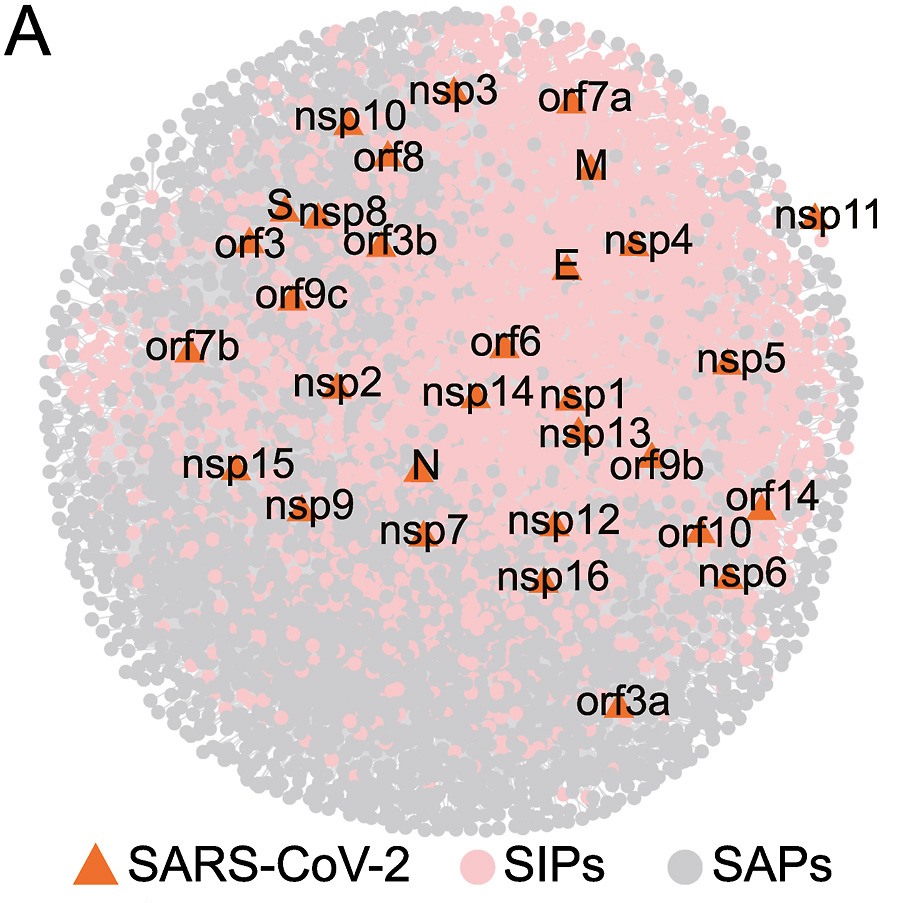
The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has seriously threatened global public health and caused huge economic losses. Omics studies of SARS-CoV-2 can help understand the interaction between the virus and host, thereby providing a new perspective in guiding the intervention and treatment of the SARS-CoV-2 infection. Since large amount of SARS-CoV-2 omics data have been accumulated in public databases, this study aimed to identify key host factors involved in SARS-CoV-2 infection through systematic integration of transcriptome and interactome data. By manually curating published studies, we obtained a comprehensive SARS-CoV-2-human protein-protein interactions (PPIs) network, comprising 3591 human proteins interacting with 31 SARS-CoV-2 viral proteins. Using the RobustRankAggregation method, we identified 123 multiple cell line common genes (CLCGs), of which 115 up-regulated CLCGs showed host enhanced innate immunity and chemotactic response signatures. Combined with network analysis, co-expression and functional enrichment analysis, we discovered four key host factors involved in SARS-CoV-2 infection: IFITM1, SERPINE1, DDX60, and TNFAIP2. Furthermore, SERPINE1 was found to facilitate SARS-CoV-2 replication, and can alleviate the endoplasmic reticulum (ER) stress induced by ORF8 protein through interaction with ORF8. Our findings highlight the importance of systematic integration analysis in understanding SARS-CoV-2-human interactions and provide valuable insights for future research on potential therapeutic targets against SARS-CoV-2 infection.
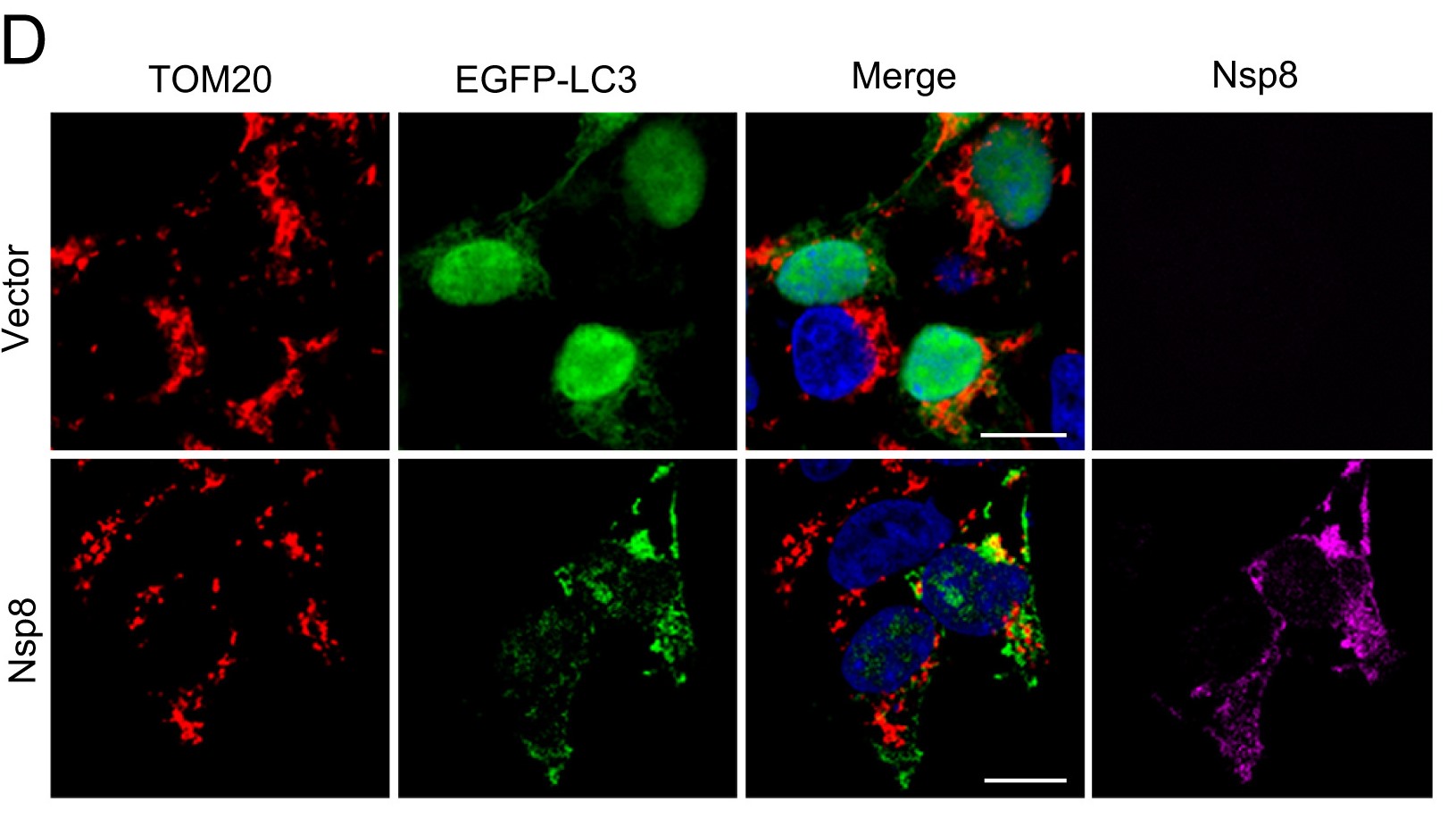
Autophagy plays an important role in the interaction between viruses and host cells. SARS-CoV-2 infection can disrupt the autophagy process in target cells. However, the precise molecular mechanism is still unknown. In this study, we discovered that the Nsp8 of SARS-CoV-2 could cause an increasing accumulation of autophagosomes by preventing the fusion of autophagosomes and lysosomes. From further investigation, we found that Nsp8 was present on mitochondria and can damage mitochondria to initiate mitophagy. The results of experiments with immunofluorescence revealed that Nsp8 induced incomplete mitophagy. Moreover, both domains of Nsp8 orchestrated their function during Nsp8-induced mitophagy, in which the N-terminal domain colocalized with mitochondria and the C-terminal domain induced auto/mitophagy. This novel finding expands our understanding of the function of Nsp8 in promoting mitochondrial damage and inducing incomplete mitophagy, which helps us to understand the etiology of COVID-19 as well as open up new pathways for creating SARS-CoV-2 treatment methods.
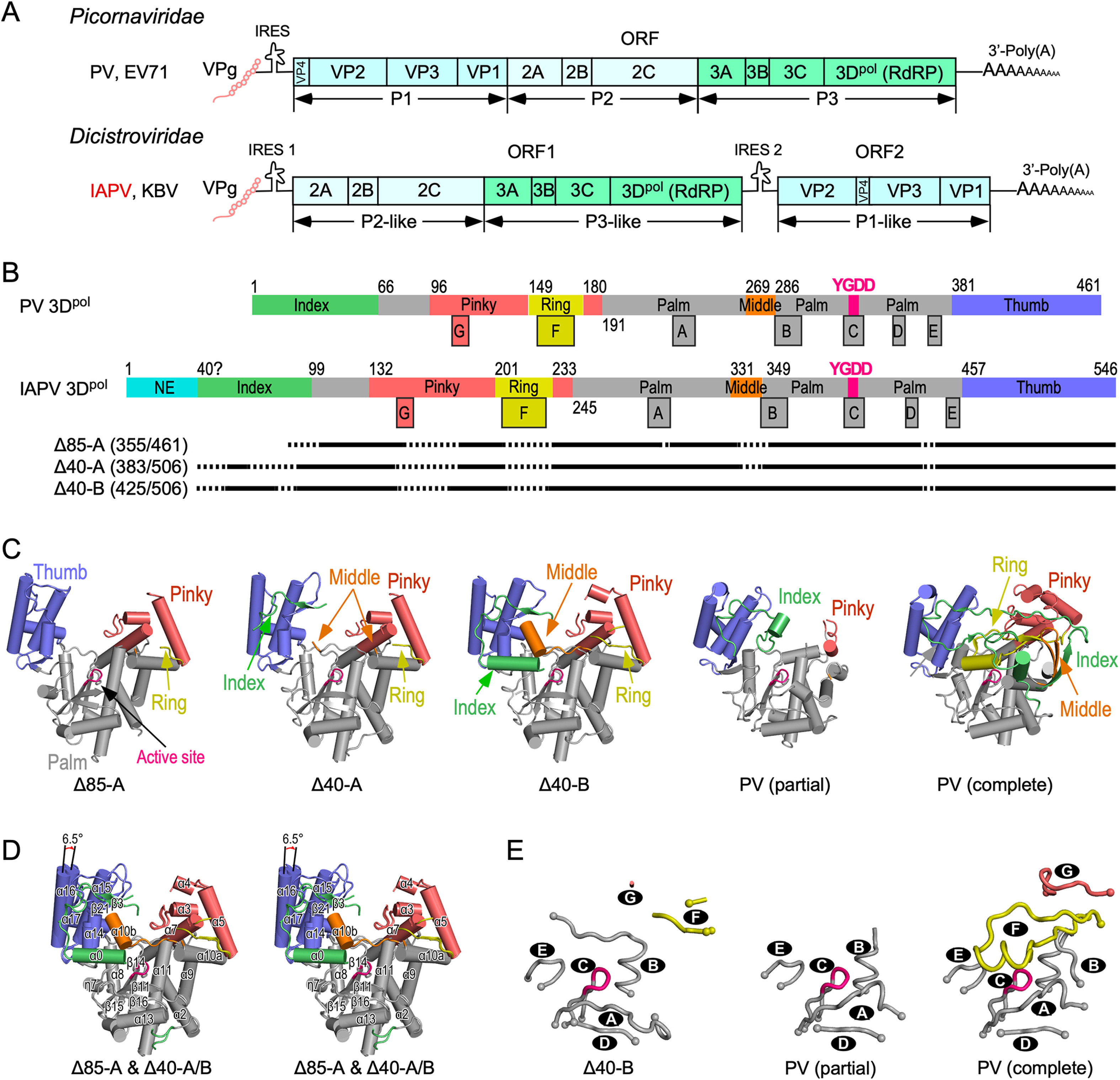
The Dicistroviridae is a virus family that includes many insect pathogens. These viruses contain a positive-sense RNA genome that is replicated by the virally encoded RNA-dependent RNA polymerase (RdRP) also named 3Dpol. Compared with the Picornaviridae RdRPs such as poliovirus (PV) 3Dpol, the Dicistroviridae representative Israeli acute paralysis virus (IAPV) 3Dpol has an additional N-terminal extension (NE) region that is about 40-residue in length. To date, both the structure and catalytic mechanism of the Dicistroviridae RdRP have remain elusive. Here we reported crystal structures of two truncated forms of IAPV 3Dpol, namely Δ85 and Δ40, both missing the NE region, and the 3Dpol protein in these structures exhibited three conformational states. The palm and thumb domains of these IAPV 3Dpol structures are largely consistent with those of the PV 3Dpol structures. However, in all structures, the RdRP fingers domain is partially disordered, while different conformations of RdRP substructures and interactions between them are also present. In particular, a large-scale conformational change occurred in the motif B-middle finger region in one protein chain of the Δ40 structure, while a previously documented alternative conformation of motif A was observed in all IAPV structures. These experimental data on one hand show intrinsic conformational variances of RdRP substructures, and on the other hand suggest possible contribution of the NE region in proper RdRP folding in IAPV.
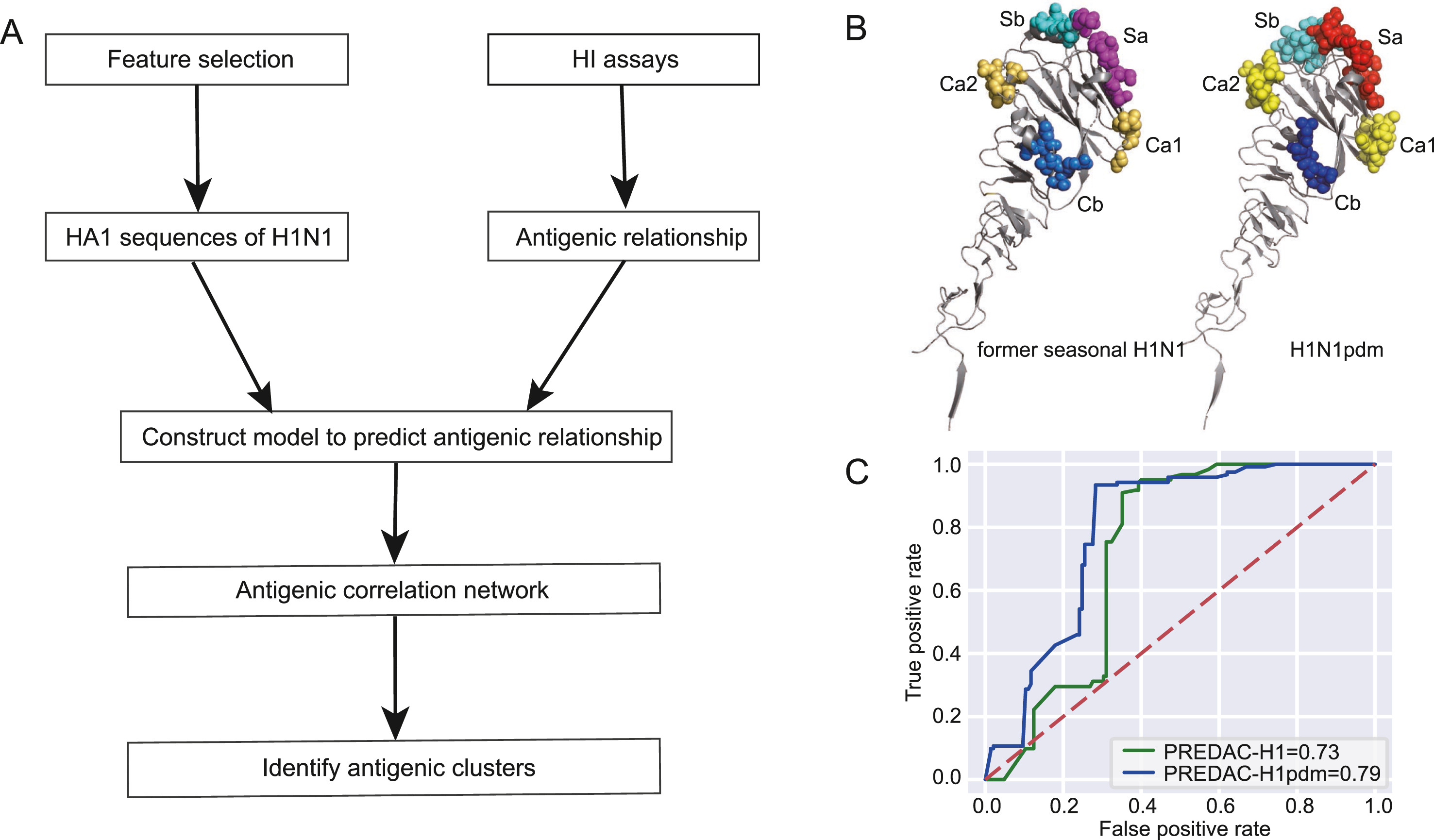
The Influenza A (H1N1) pdm09 virus caused a global pandemic in 2009 and has circulated seasonally ever since. As the continual genetic evolution of hemagglutinin in this virus leads to antigenic drift, rapid identification of antigenic variants and characterization of the antigenic evolution are needed. In this study, we developed PREDAC-H1pdm, a model to predict antigenic relationships between H1N1pdm viruses and identify antigenic clusters for post-2009 pandemic H1N1 strains. Our model performed well in predicting antigenic variants, which was helpful in influenza surveillance. By mapping the antigenic clusters for H1N1pdm, we found that substitutions on the Sa epitope were common for H1N1pdm, whereas for the former seasonal H1N1, substitutions on the Sb epitope were more common in antigenic evolution. Additionally, the localized epidemic pattern of H1N1pdm was more obvious than that of the former seasonal H1N1, which could make vaccine recommendation more sophisticated. Overall, the antigenic relationship prediction model we developed provides a rapid determination method for identifying antigenic variants, and the further analysis of evolutionary and epidemic characteristics can facilitate vaccine recommendations and influenza surveillance for H1N1pdm.
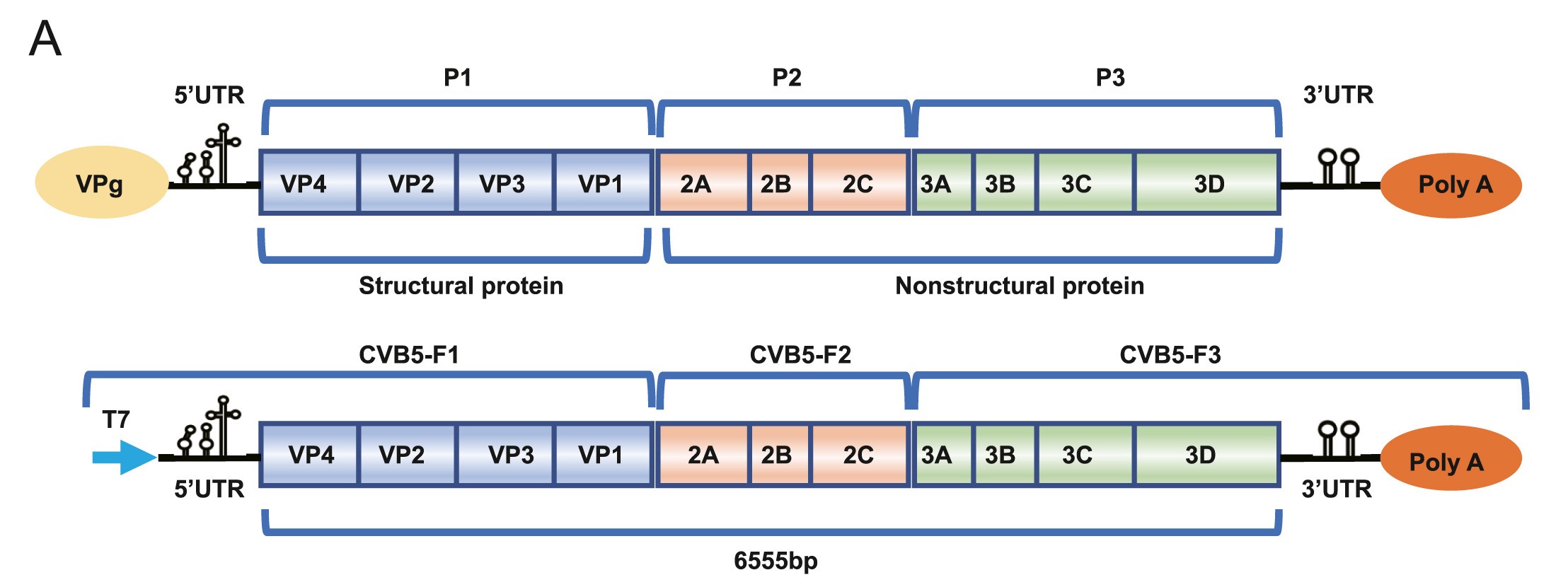
Coxsackievirus belongs to the Picornaviridae family and is one of the major pathogens that cause hand, foot and mouth disease (HFMD) in infants and children with potential serious complications and even deaths. The pathogenesis of this virus is not fully elucidated and no vaccine or antiviral drug has been approved. In this study, a full-length infectious cDNA clone of coxsackievirus B5 virus was assembled and the recombinant virus displayed similar growth kinetics and ability to cause cytopathic effects as the parental virus. Luciferase reporter was then incorporated to generate both full-length and subgenomic replicon (SGR) reporter viruses. The full-length reporter virus is suitable for high-throughput antiviral screening, while the SGR is a useful tool to study viral-host interactions. More importantly, the full-length reporter virus has also been shown to infect the suckling mouse model and the reporter gene could be detected using an in vivo imaging system, thus providing a powerful tool to track viruses in vivo. In summary, we have generated coxsackievirus B5 reporter viruses and provided unique tools for studying virus-host interactions in vitro and in vivo as well as for high-throughput screenings (HTS) to identify novel antivirals.
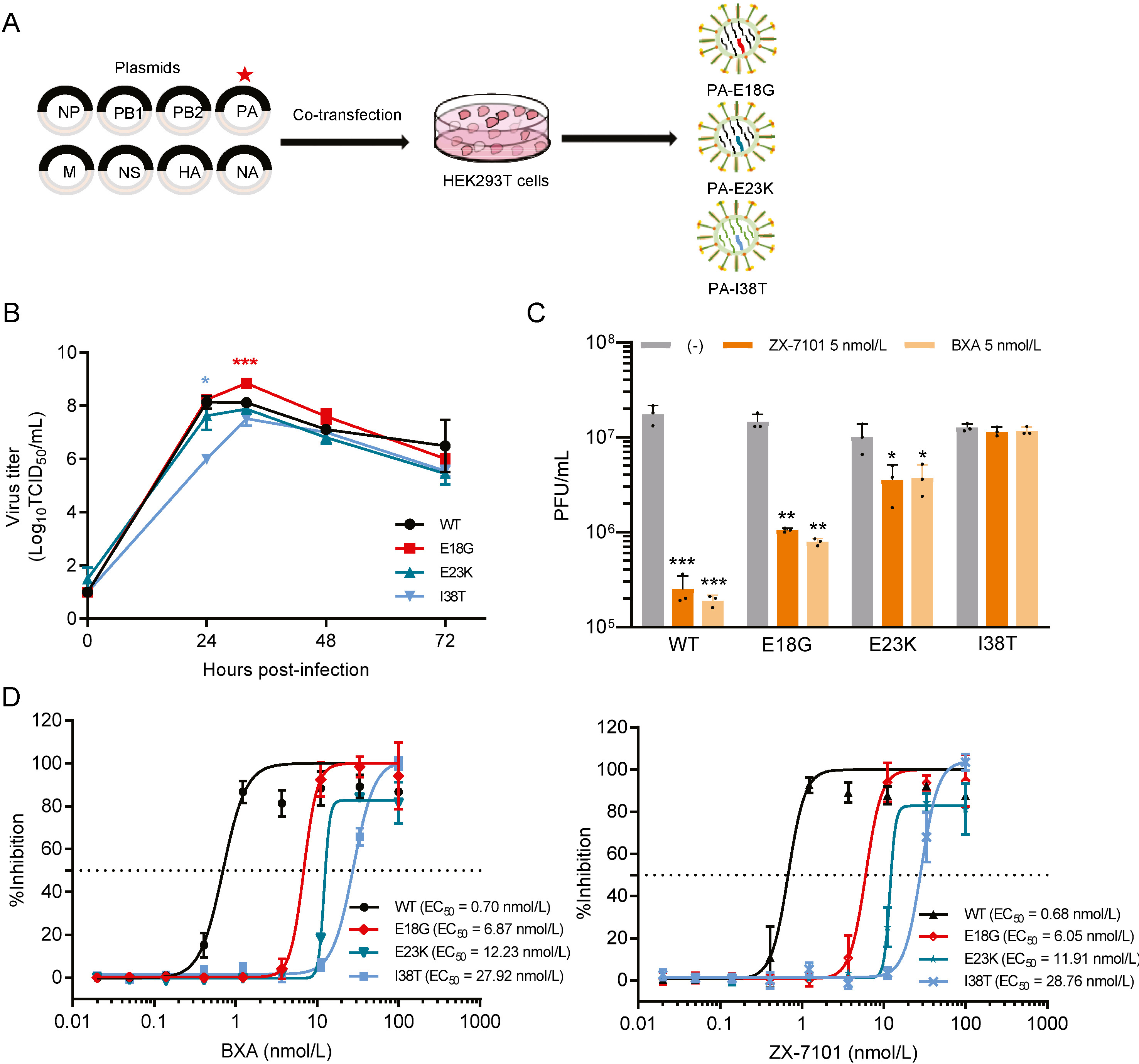
Cap-dependent endonuclease (CEN) in the polymerase acidic protein (PA) of influenza A virus (IAV) represents a promising drug target due to its critical role in viral gene transcription. The CEN inhibitor, baloxavir marboxil (BXM), was approved in Japan and the US in 2018 and several other countries subsequently. Along with the clinical use of BXM, the emergence and spread of IAV variants with reduced susceptibility to BXM have aroused serious concern. Herein, we comprehensively characterized the in vitro and in vivo antiviral activities of ZX-7101A, an analogue of BXM. The active form of prodrug ZX-7101 showed broad-spectrum antiviral potency against various IAV subtypes, including pH1N1, H3N2, H7N9 and H9N2, in MDCK cells, and the 50% effective concentration (EC50) was calculated to nanomole level and comparable to that of baloxavir acid (BXA), the active form of BXM. Furthermore, in vivo assays showed that administration of ZX-7101A conferred significant protection against lethal pH1N1 challenge in mice, with reduced viral RNA loads and alleviated pulmonary damage. Importantly, serial passaging of H1N1 virus in MDCK cells under selection pressure of ZX-7101 led to a resistant variant at the 15th passage. Reverse genetic and sequencing analysis demonstrated that a single E18G substitution in the PA subunit contributed to the reduced susceptibility to both ZX-7101 and BXA. Taken together, our results not only characterized a new CEN inhibitor of IAV but also identified a novel amino acid substitution responsible for CEN inhibitor resistance, which provides critical clues for future drug development and drug resistance surveillance.
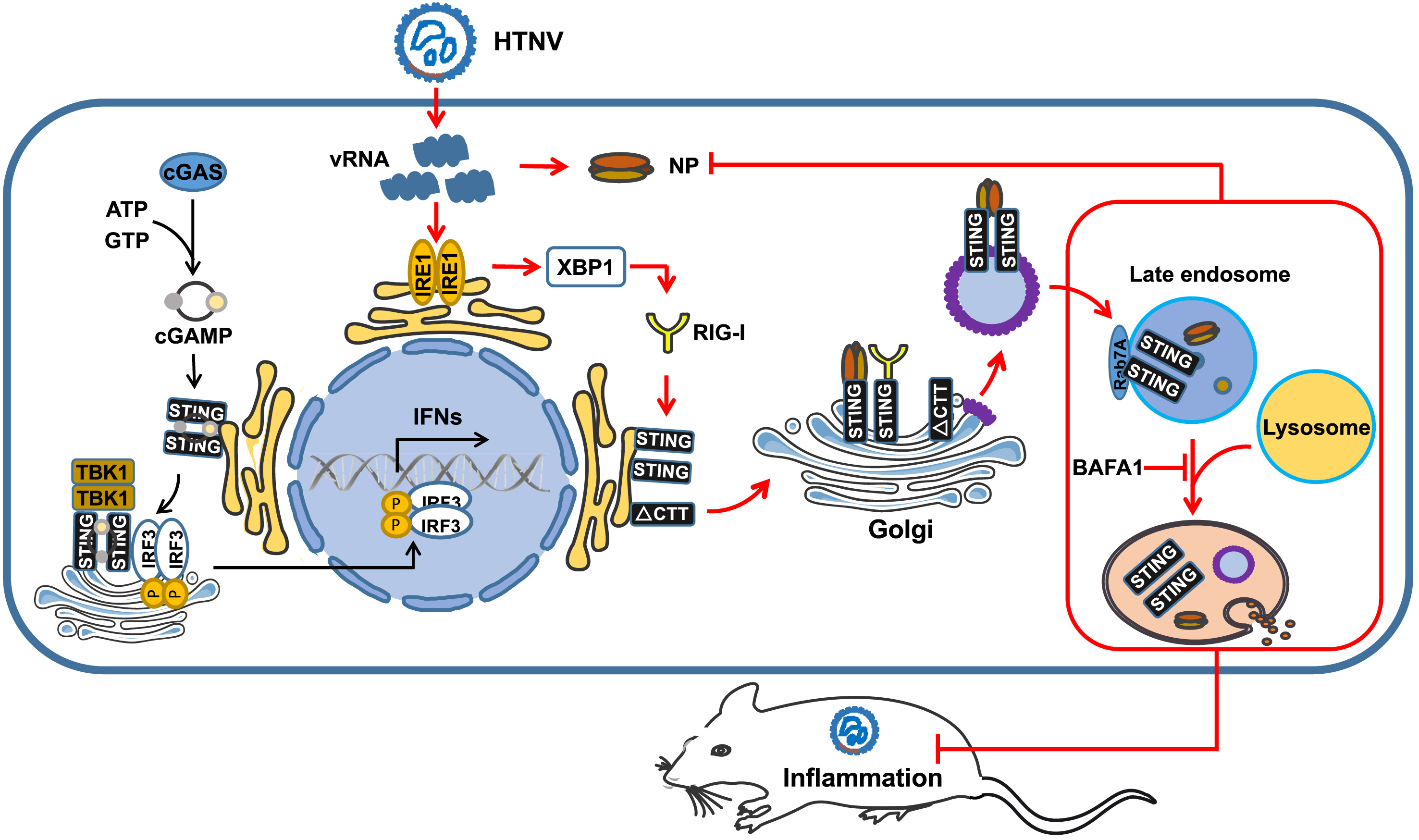
Hantaan virus (HTNV), the prototype virus of hantavirus, could escape innate immunity by restraining type I interferon (IFN) responses. It is largely unknown whether there existed other efficient anti-hantaviral tactics in host cells. Here, we demonstrate that the stimulator of interferon genes (STING) strengthens the host IFN-independent anti-hantaviral immunity. HTNV infection activates RIG-I through IRE1-XBP 1-mediated ER stress, which further facilitates the subcellular translocation and activation of STING. During this process, STING triggers cellular autophagy by interacting with Rab7A, thus restricting viral replication. To note, the anti-hantaviral effects of STING are independent of canonical IFN signaling. Additionally, neither application of the pharmacological antagonist nor the agonist targeting STING could improve the outcomes of nude mice post HTNV challenge in vivo. However, the administration of plasmids exogenously expressing the mutant C-terminal tail (ΔCTT) STING, which would not trigger the type I IFN responses, protected the nude mice from lethal HTNV infection. In summary, our research revealed a novel antiviral pathway through the RIG-I-STING-autophagy pathway, which offered novel therapeutic strategies against hantavirus infection.
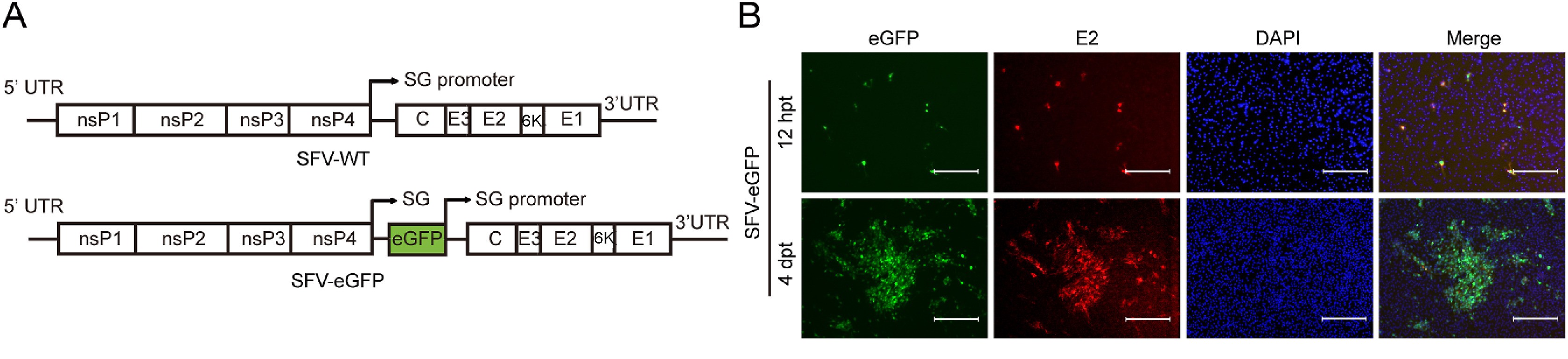
Alphaviruses, which contain a variety of mosquito-borne pathogens, are important pathogens of emerging/re-emerging infectious diseases and potential biological weapons. Currently, no specific antiviral drugs are available for the treatment of alphaviruses infection. For most highly pathogenic alphaviruses are classified as risk group-3 agents, the requirement of biosafety level 3 (BSL-3) facilities limits the live virus-based antiviral study. To facilitate the antiviral development of alphaviruses, we developed a high throughput screening (HTS) platform based on a recombinant Semliki Forest virus (SFV) which can be manipulated in BSL-2 laboratory. Using the reverse genetics approach, the recombinant SFV and SFV reporter virus expressing eGFP (SFV-eGFP) were successfully rescued. The SFV-eGFP reporter virus exhibited robust eGFP expression and remained relatively stable after four passages in BHK-21 cells. Using a broad-spectrum alphavirus inhibitor ribavirin, we demonstrated that the SFV-eGFP can be used as an effective tool for antiviral study. The SFV-eGFP reporter virus-based HTS assay in a 96-well format was then established and optimized with a robust Z′ score. A section of reference compounds that inhibit highly pathogenic alphaviruses were used to validate that the SFV-eGFP reporter virus-based HTS assay enables rapid screening of potent broad-spectrum inhibitors of alphaviruses. This assay provides a safe and convenient platform for antiviral study of alphaviruses.
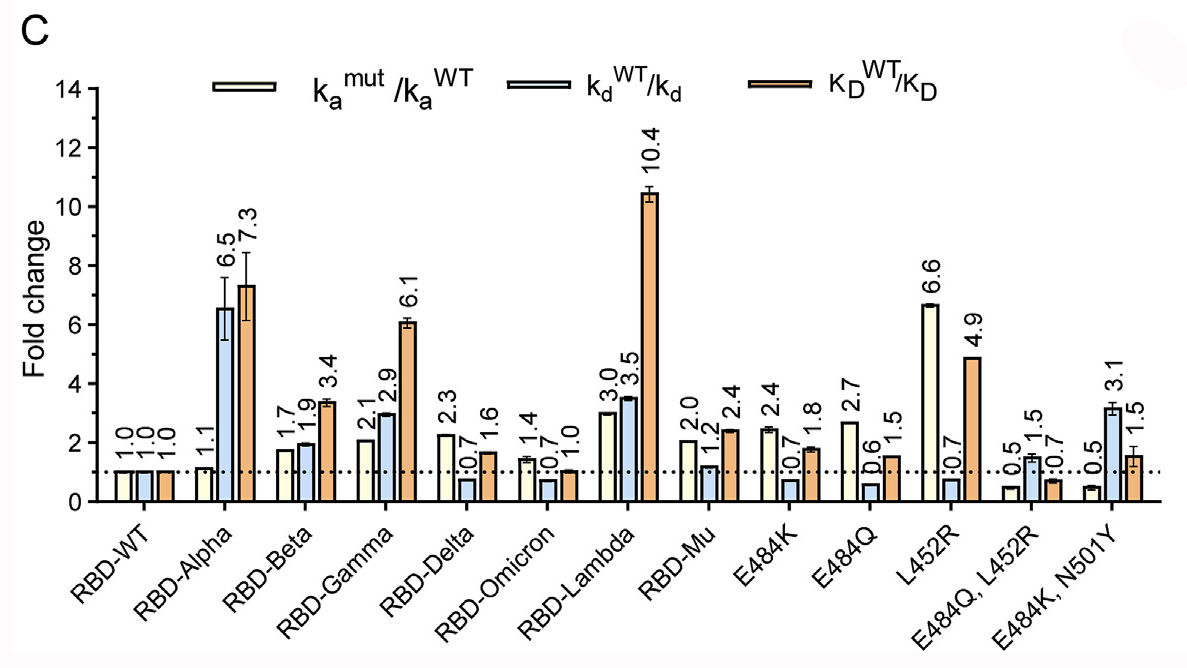
SARS-CoV-2 variants are constantly emerging, hampering public health measures in controlling the number of infections. While it is well established that mutations in spike proteins observed for the different variants directly affect virus entry into host cells, there remains a need for further expansion of systematic and multifaceted comparisons. Here, we comprehensively studied the effect of spike protein mutations on spike expression and proteolytic activation, binding affinity, viral entry efficiency and host cell tropism of eight variants of concern (VOC) and variants of interest (VOI). We found that both the full-length spike and its receptor-binding domain (RBD) of Omicron bind to hACE2 with an affinity similar to that of the wild-type. In addition, Alpha, Beta, Delta and Lambda pseudoviruses gained significantly enhanced cell entry ability compared to the wild-type, while the Omicron pseudoviruses showed a slightly increased cell entry, suggesting the vastly increased rate of transmission observed for Omicron variant is not associated with its affinity to hACE2. We also found that the spikes of Omicron and Mu showed lower S1/S2 cleavage efficiency and inefficiently utilized TMPRSS2 to enter host cells than others, suggesting that they prefer the endocytosis pathway to enter host cells. Furthermore, all variants' pseudoviruses we tested gained the ability to enter the animal ACE2-expressing cells. Especially the infection potential of rats and mice showed significantly increased, strongly suggesting that rodents possibly become a reservoir for viral evolution. The insights gained from this study provide valuable guidance for a targeted approach to epidemic control, and contribute to a better understanding of SARS-CoV-2 evolution.
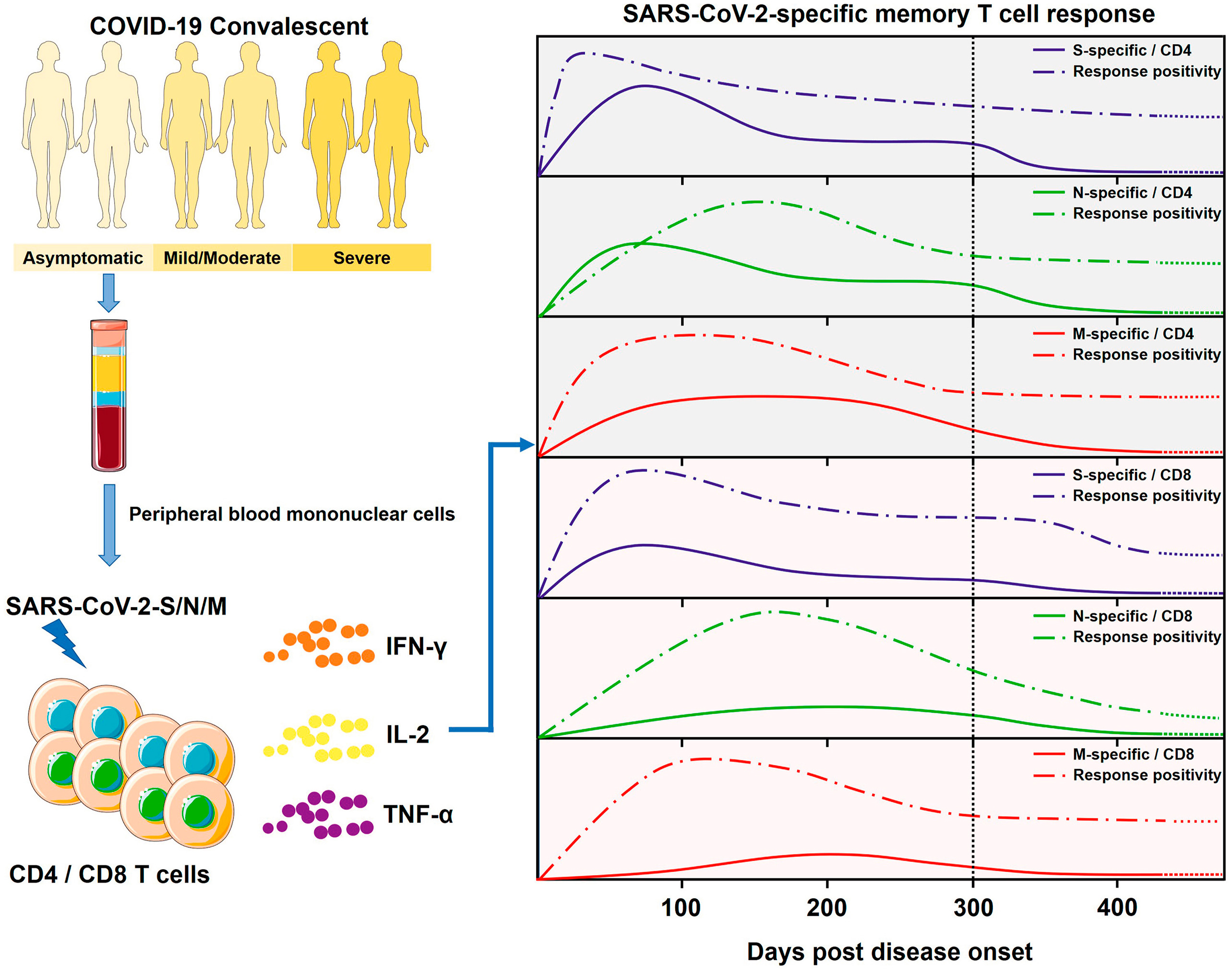
A key question in the coronavirus disease 2019 (COVID-19) pandemic is the duration of specific T cell responses against the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) post primary infection, which is difficult to address due to the large-scale COVID-19 vaccination and re-exposure to the virus. Here, we conducted an analysis of the long-term SARS-CoV-2-specific T cell responses in a unique cohort of convalescent individuals (CIs) that were among the first to be infected worldwide and without any possible antigen re-exposure since then. The magnitude and breadth of SARS-CoV-2-specific T cell responses correlated inversely with the time that had elapsed from disease onset and the age of those CIs. The mean magnitude of SARS-CoV-2-specific CD4 and CD8 T cell responses decreased about 82% and 76%, respectively, over the time period of ten months after infection. Accordingly, the longitudinal analysis also demonstrated that SARS-CoV-2-specific T cell responses waned significantly in 75% of CIs during the follow-up. Collectively, we provide a comprehensive characterization of the long-term memory T cell response in CIs, suggesting that robust SARS-CoV-2-specific T cell immunity post primary infection may be less durable than previously expected.
A rapid and accurate COVID-19 diagnosis is a prerequisite for blocking the source of infection as soon as possible and taking the appropriate medical action. Herein, we developed GeneClick, a device for nucleic acid self-testing of SARS-CoV-2, consisting of three modules: a sampling kit, a microfluidic chip-based disposable cartridge, and an amplification reader. In addition, we evaluated the clinical performance of GeneClick using 2162 nasal swabs collected at three medical institutions, using three commercial RT-qPCR kits and an antigen self-test as references. Compared to RT-qPCR, the sensitivity and specificity of the GeneClick assay were 97.93% and 99.72%, respectively, with a kappa value of 0.979 (P < 0.01). Of the 2162 samples, 2076 were also tested for SARS-CoV-2 antigens. Among the 314 positive samples identified by GeneClick assay, 63 samples were undetected by antigen tests. Overall, the GeneClick nucleic acid self-test demonstrated higher accuracy than the antigen-based detection. Based on the additional features, including simple operation, affordable price, portable device, and reliability of smartphone APP-driven sampling and result reporting, GeneClick offers a powerful tool for field-based SARS-CoV-2 detection in primary healthcare institutions or at-home use.
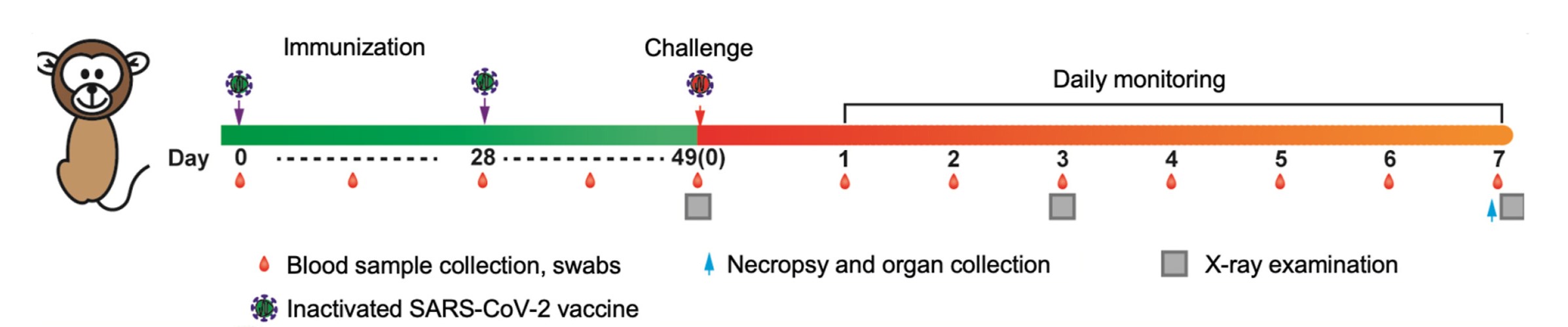
Highlights 1. The Delta inactivated vaccine constructed in this study has high immunogenicity and good safety. 2. The inactivated vaccine has good cross-protection against SARS-CoV-2 variants. 3. The vaccine exhibited high productivity and good genetic stability in production.
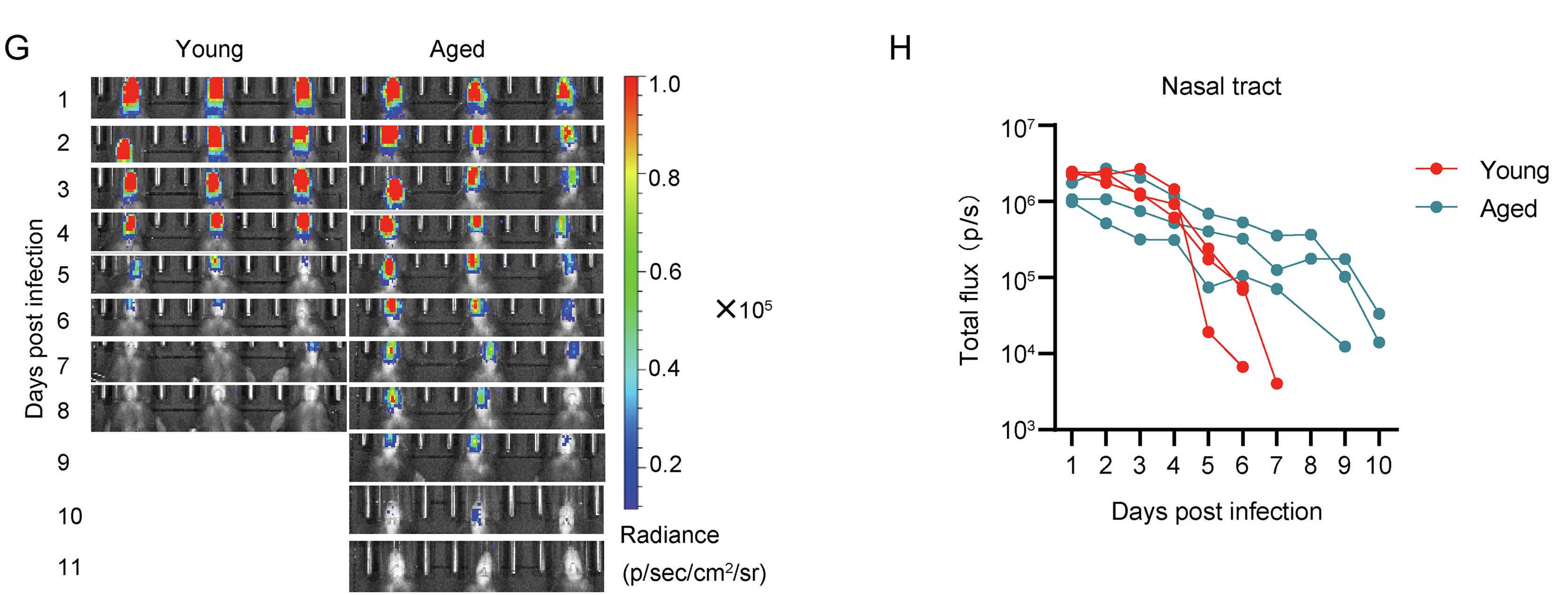
Highlights 1. The in vivo BLI model of IAV infections can simplify the determination of viral load in living animals; 2. The in vivo BLI model of IAV infections allow longitudinal measurements of virus infection/spread in living animals; 3. The in vivo BLI model of IAV infections improved the throughput of animal models; 4. The advanced BLI models can facilitate studies in both basic and applied virology.
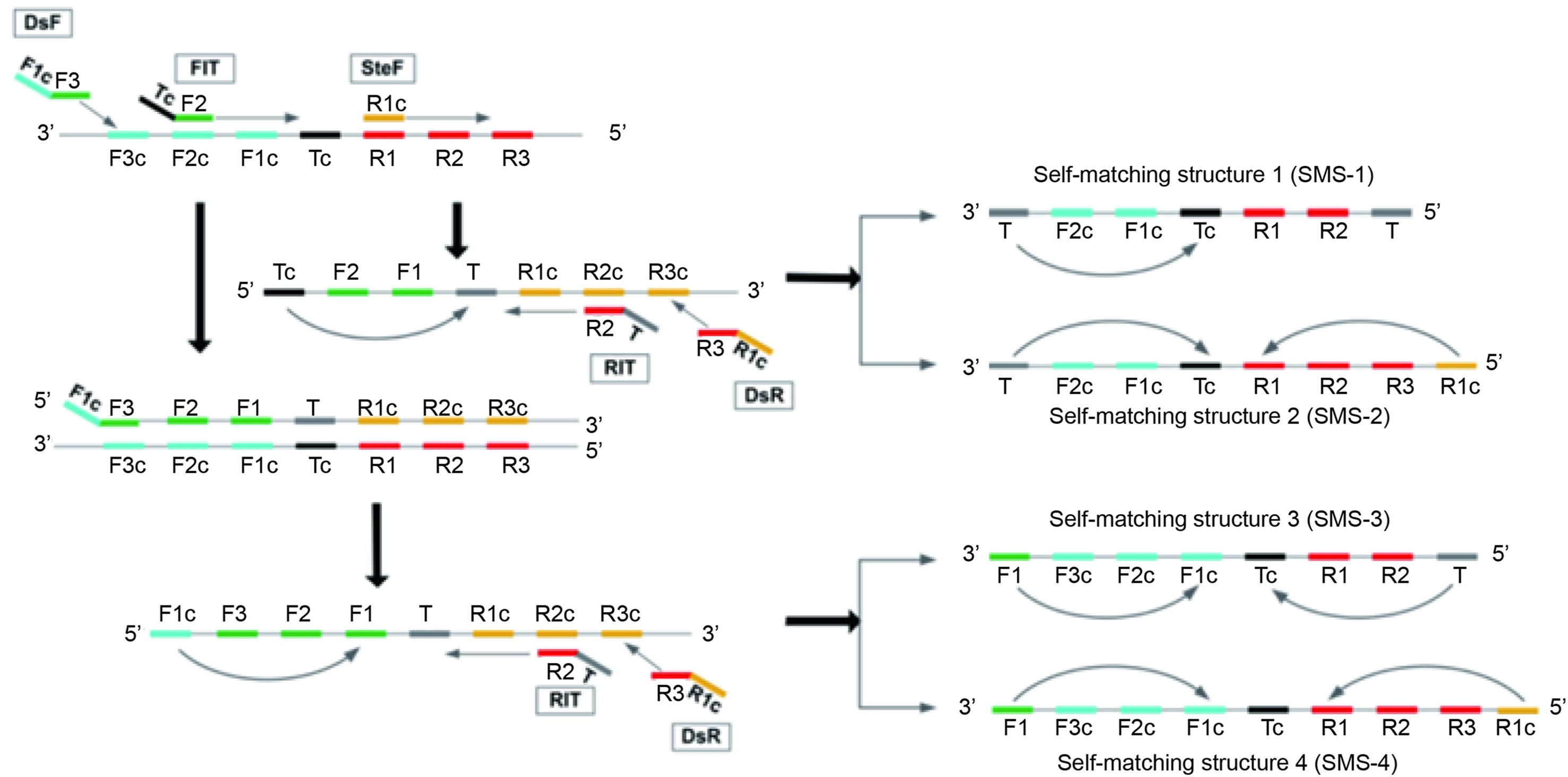
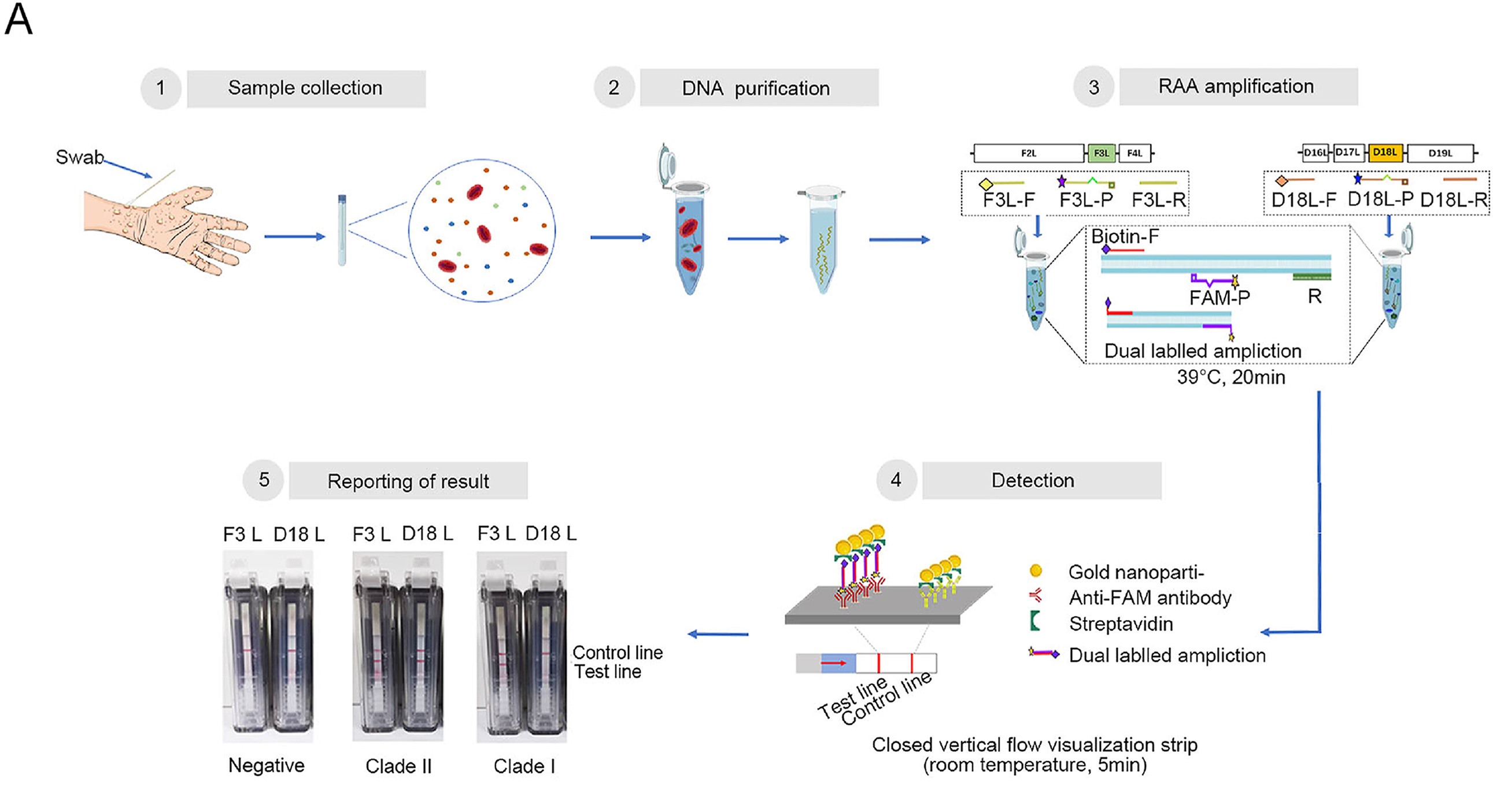
Highlights 1. The newly developed visual assay panel is a rapid and reliable tool to differentiate the clade I and clade II MPXV within 25 min. 2. The panel combines recombinase-aid amplification and immunochromatography, and detects as low as 1 copy/μL recombinant plasmid. 3. Visual assay panel shows no cross-reactivity with orthopoxviruses and herpesvirus that infect humans, such as vaccinia virus.
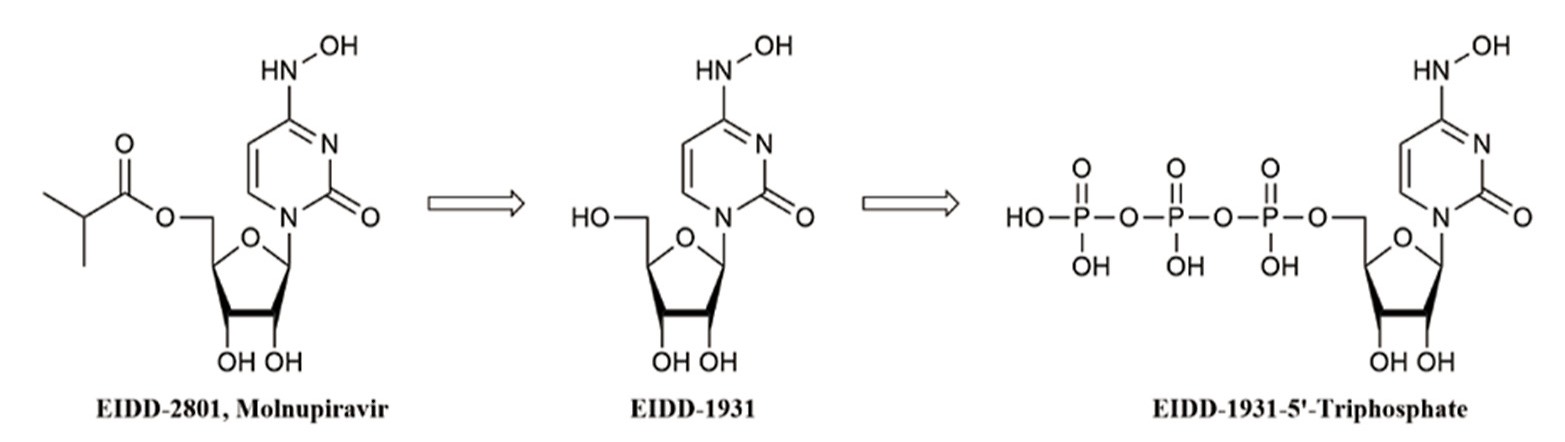
Highlights 1. Molnupiravir exhibits effective antiviral activity against ZIKV in vitro. 2. Intraperitoneal administration of Molnupiravir protects mice from lethal ZIKV challenge. 3. Molnupiravir might act on the replication phase of the ZIKV life cycle.
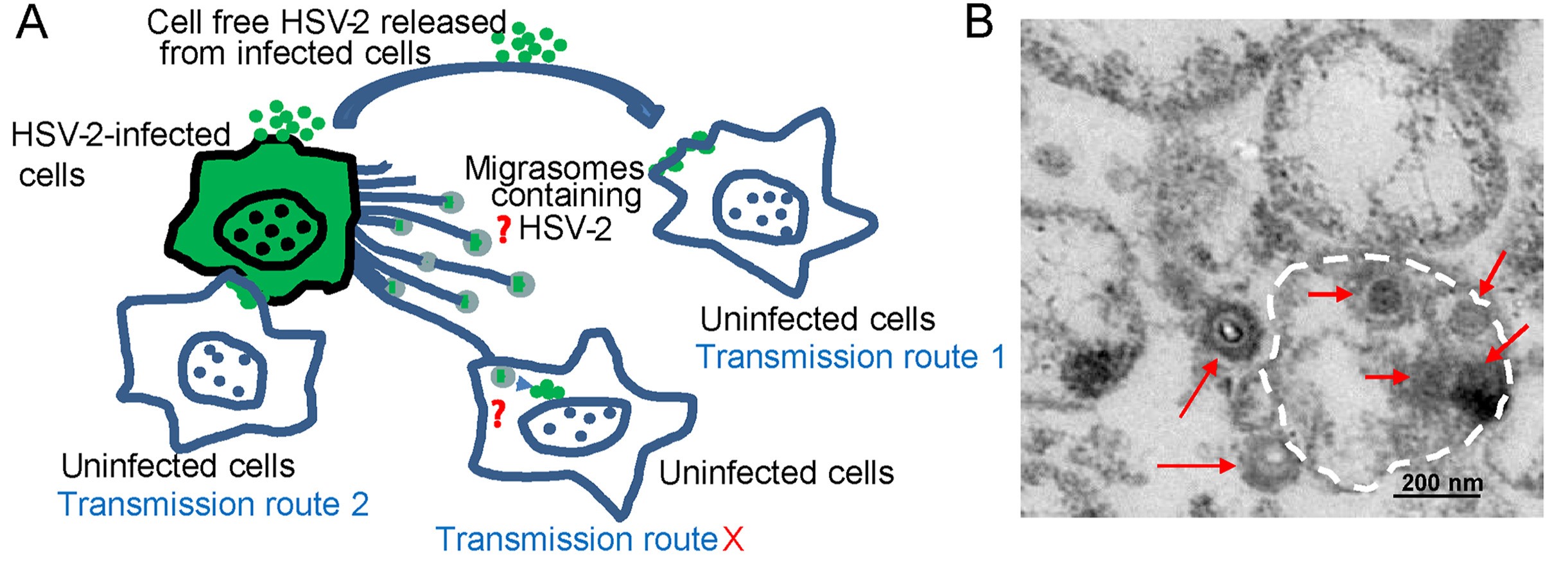
Highlights 1. Cells infected with HSV-2 release migrasomes containing HSV-2 virions. 2. HSV-2 in the isolated migrasomes can be transmitted to uninfected cells and cause productive infection. 3.It is the first time that migrasomes have been found to play a role in virus spread.
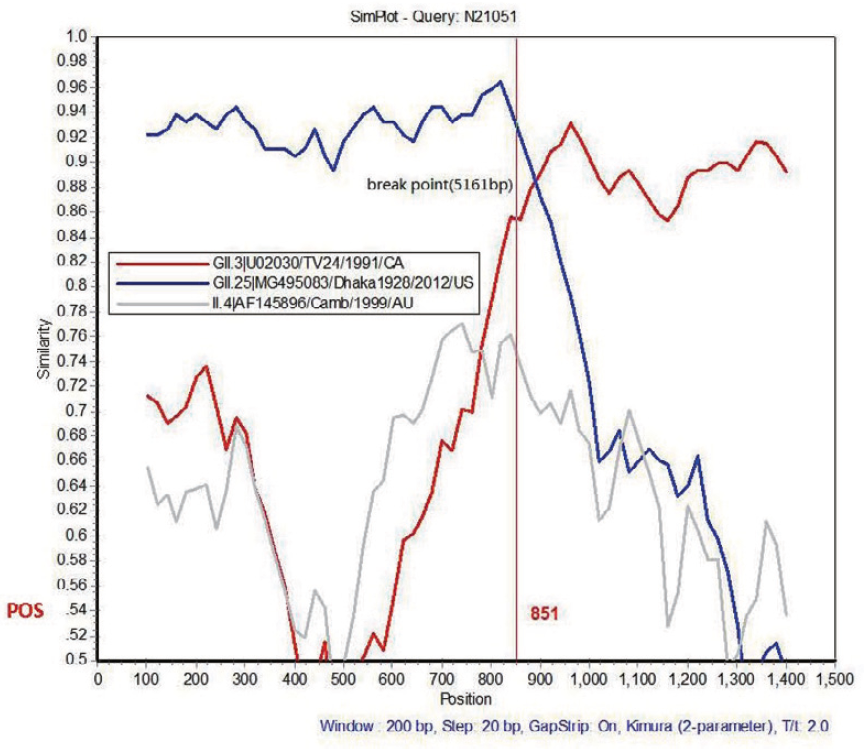
Highlights 1. The first genome of GII.3[P25] strain isolated in China was determined by NGS. 2. About 19 unique amino acid substitutions were identified in the VP1 region of GII.3[P25]. 3. Antigenic variation may have contributed to the re-emerge of GII.3[P25] strains.